

Neurogenic inflammation, or NI, is the physiological process where mediators are discharged directly from the cutaneous nerves to commence an inflammatory response. This results in the creation of local inflammatory reactions including, erythema, swelling, temperature increase, tenderness, and pain. Fine unmyelinated afferent somatic C-fibers, which respond to low intensity mechanical and chemical stimulations, are largely responsible for the release of these inflammatory mediators.
When stimulated, these nerve pathways in the cutaneous nerves release energetic neuropeptides, or substance P and calcitonin gene related peptide (CGRP), rapidly into the microenvironment, triggering a series of inflammatory responses. There is a significant distinction in immunogenic inflammation, that’s the very first protective and reparative response made by the immune system when a pathogen enters the body, whereas neurogenic inflammation involves a direct connection between the nervous system and the inflammatory responses. Even though neurogenic inflammation and immunologic inflammation can exist concurrently, the two are not clinically indistinguishable. The purpose of the article below is to discuss the mechanism of neurogenic inflammation and the peripheral nervous system’s role in host defense and immunopathology.
Table of Contents
Neurogenic Inflammation – The Peripheral Nervous System’s Role in Host Defense and Immunopathology
Abstract
The peripheral nervous and immune systems are traditionally thought of as serving separate functions. This line is, however, becoming increasingly blurred by new insights into neurogenic inflammation. Nociceptor neurons possess many of the same molecular recognition pathways for danger as immune cells and in response to danger, the peripheral nervous system directly communicates with the immune system, forming an integrated protective mechanism. The dense innervation network of sensory and autonomic fibers in peripheral tissues and high speed of neural transduction allows for rapid local and systemic neurogenic modulation of immunity. Peripheral neurons also appear to play a significant role in immune dysfunction in autoimmune and allergic diseases. Therefore, understanding the coordinated interaction of peripheral neurons with immune cells may advance therapeutic approaches to increase host defense and suppress immunopathology.
Introduction
Two thousand years ago, Celsus defined inflammation as involving four cardinal signs – Dolor (pain), Calor (heat), Rubor (redness), and Tumor (swelling), an observation indicating that activation of the nervous system was recognized as being integral to inflammation. However, pain has been mainly thought of since then, only as a symptom, and not a participant in the generation of inflammation. In this perspective, we show that the peripheral nervous system plays a direct and active role in modulating innate and adaptive immunity, such that the immune and nervous systems may have a common integrated protective function in host defense and the response to tissue injury, an intricate interaction that also can lead to pathology in allergic and autoimmune diseases.
Survival of organisms is critically dependent on the capacity to mount a defense against potential harm from tissue damage and infection. Host defense involves both avoidance behavior to remove contact with a dangerous (noxious) environment (a neural function), and active neutralization of pathogens (an immune function). Traditionally, the role of the immune system in combating infective agents and repairing tissue injury has been considered quite distinct from that of the nervous system, which transduces damaging environmental and internal signals into electrical activity to produce sensations and reflexes (Fig. 1). We propose that these two systems are actually components of a unified defense mechanism. The somatosensory nervous system is ideally placed to detect danger. Firstly, all tissues that are highly exposed to the external environment, such as epithelial surfaces of the skin, lungs, urinary and digestive tract, are densely innervated by nociceptors, high threshold pain-producing sensory fibers. Secondly, transduction of noxious external stimuli is almost instantaneous, orders of magnitude quicker than the mobilization of the innate immune system, and therefore may be the “first responder” in host defense.
In addition to orthodromic inputs to the spinal cord and brain from the periphery, action potentials in nociceptor neurons can also be transmitted antidromically at branch points back down to the periphery, the axon reflex. These together with sustained local depolarizations lead to a rapid and local release of neural mediators from both peripheral axons and terminals (Fig. 2) 1. Classic experiments by Goltz (in 1874) and by Bayliss (in 1901) showed that electrically stimulating dorsal roots induces skin vasodilation, which led to the concept of a “neurogenic inflammation”, independent of that produced by the immune system (Fig. 3).

Neurogenic inflammation is mediated by the release of the neuropeptides calcitonin gene related peptide (CGRP) and substance P (SP) from nociceptors, which act directly on vascular endothelial and smooth muscle cells 2–5. CGRP produces vasodilation effects 2, 3, whereas SP increases capillary permeability leading to plasma extravasation and edema 4, 5, contributing to the rubor, calor and tumor of Celsus. However, nociceptors release many additional neuropeptides (online database: http://www.neuropeptides.nl/), including Adrenomedullin, Neurokinins A and B, Vasoactive intestinal peptide (VIP), neuropeptide (NPY), and gastrin releasing peptide (GRP), as well as other molecular mediators such as glutamate, nitric oxide (NO) and cytokines such as eotaxin 6.
We now appreciate that the mediators released from sensory neurons in the periphery not only act on the vasculature, but also directly attract and activate innate immune cells (mast cells, dendritic cells), and adaptive immune cells (T lymphocytes) 7–12. In the acute setting of tissue damage, we conjecture that neurogenic inflammation is protective, facilitating physiological wound healing and immune defense against pathogens by activating and recruiting immune cells. However, such neuro-immune communications also likely play major roles in the pathophysiology of allergic and autoimmune diseases by amplifying pathological or maladaptive immune responses. In animal models of rheumatoid arthritis for example, Levine and colleagues have shown that denervation of the joint leads to a striking attenuation in inflammation, that is dependent on neural expression of substance P 13, 14. In recent studies of allergic airway inflammation, colitis and psoriasis, primary sensory neurons play a central role in initiating and augmenting the activation of innate and adaptive immunity 15–17.
We propose therefore, that the peripheral nervous system not only plays a passive role in host defense (detection of noxious stimuli and initiation of avoidance behavior), but also an active role in concert with the immune system in modulating the responses to and combat of harmful stimuli, a role that can be subverted to contribute to disease.
Shared Danger Recognition Pathways in the Peripheral Nervous and Innate Immune Systems
Peripheral sensory neurons are adapted to recognize danger to the organism by virtue of their sensitivity to intense mechanical, thermal and irritant chemical stimuli (Fig. 1). Transient receptor potential (TRP) ion channels are the most widely studied molecular mediators of nociception, conducting non-selective entry of cations upon activation by various noxious stimuli. TRPV1 is activated by high temperatures, low pH and capsaicin, the vallinoid irritant component of chili peppers 18. TRPA1 mediates the detection of reactive chemicals including environmental irritants such as tear gas and industrial isothiocyanates 19, but more importantly, it is also activated during tissue injury by endogenous molecular signals including 4-hydroxynonenal and prostaglandins 20, 21.
Interestingly, sensory neurons share many of the same pathogen and danger molecular recognition receptor pathways as innate immune cells, which enable them also to detect pathogens (Fig. 1). In the immune system, microbial pathogens are detected by germline encoded pattern recognition receptors (PRRs), which recognize broadly conserved exogenous pathogen-associated molecular patterns (PAMPs). The first PRRs to be identified were members of toll-like receptor (TLR) family, which bind to yeast, bacterial derived cell-wall components and viral RNA 22. Following PRR activation, downstream signaling pathways are turned on that induce cytokine production and activation of adaptive immunity. In addition to TLRs, innate immune cells are activated during tissue injury by endogenous derived danger signals, also known as damage-associated molecular patterns (DAMPs) or alarmins 23, 24. These danger signals include HMGB1, uric acid, and heat shock proteins released by dying cells during necrosis, activating immune cells during non-infectious inflammatory responses.
PRRs including TLRs 3, 4, 7, and 9 are expressed by nociceptor neurons, and stimulation by TLR ligands leads to induction of inward currents and sensitization of nociceptors to other pain stimuli 25–27. Furthermore, activation of sensory neurons by the TLR7 ligand imiquimod leads to activation of an itch specific sensory pathway 25. These results indicate that infection-associated pain and itch may be partly due to direct activation of neurons by pathogen-derived factors, which in turn activate immune cells through peripheral release of neuronal signaling molecules.
A major DAMP/alarmin released during cellular injury is ATP, which is recognized by purinergic receptors on both nociceptor neurons and immune cells 28–30. Purinergic receptors are made up of two families: P2X receptors, ligand-gated cation channels, and P2Y receptors, G-protein coupled receptors. In nociceptor neurons, recognition of ATP occurs through P2X3, leading to rapidly densensitizing cation currents and pain 28, 30 (Fig. 1), while P2Y receptors contribute to nociceptor activation by sensitization of TRP and voltage-gated sodium channels. In macrophages, ATP binding to P2X7 receptors leads to hyperpolarization, and downstream activation of the inflammasome, a molecular complex important in generation of IL-1beta and IL-18 29. Therefore, ATP is a potent danger signal that activates both peripheral neurons and innate immunity during injury, and some evidence even suggests that neurons express parts of the inflammasome molecular machinery 31.
The flip side of danger signals in nociceptors is the role of TRP channels in immune cell activation. TRPV2, a homologue of TRPV1 activated by noxious heat, is expressed at high levels in innate immune cells 32. Genetic ablation of TRPV2 led to defects in macrophage phagocytosis and clearance of bacterial infections 32. Mast cells also express TRPV channels, which may directly mediate their degranulation 33. It remains to be determined whether endogenous danger signals activate immune cells in a similar manner as nociceptors.
A key means of communication between immune cells and nociceptor neurons are through cytokines. Upon activation of cytokine receptors, signal transduction pathways are activated in sensory neurons leading to downstream phosphorylation of membrane proteins including TRP and voltage-gated channels (Fig. 1). The resulting sensitization of nociceptors means that normally innocuous mechanical and heat stimuli can now activate nociceptors. Interleukin 1 beta and TNF-alpha are two important cytokines released by innate immune cells during inflammation. IL-1beta and TNF-alpha are directly sensed by nociceptors which express the cognate receptors, induce activation of p38 map kinases leading to increased membrane excitability 34–36. Nerve growth factor (NGF) and prostaglandin E(2) are also major inflammatory mediators released from immune cells that act directly on peripheral sensory neurons to cause sensitization. An important effect of nociceptor sensitization by immune factors is an increased release of neuropeptides at peripheral terminals that further activate immune cells, thereby inducing a positive feedback loop that drives and facilitates inflammation.
Sensory Nervous System Control of Innate and Adaptive Immunity
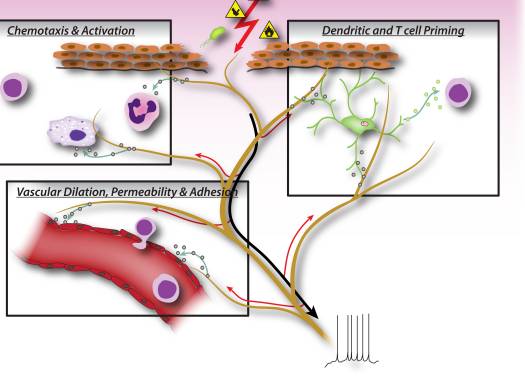
In early phases of inflammation, sensory neurons signal to tissue resident mast cells and dendritic cells, which are innate immune cells important in initiating the immune response (Fig. 2). Anatomical studies have shown a direct apposition of terminals with mast cells, as well as with dendritic cells, and the neuropeptides released from nociceptors can induce degranulation or cytokine production in these cells 7, 9, 37. This interaction plays an important role in allergic airway inflammation and dermatitis 10–12.
During the effector phase of inflammation, immune cells need to find their way to the specific site of injury. Many mediators released from sensory neurons, neuropeptides, chemokines, and glutamate, are chemotactic for neutrophils, eosinophils, macrophages, and T-cells, and enhance endothelial adhesion which facilitates immune cell homing 6, 38–41 (Fig. 2). Furthermore, some evidence implies that neurons may directly participate in the effector phase, as neuropeptides themselves may have direct antimicrobial functions 42.
Neuronally derived signaling molecules can also direct the type of inflammation, by contributing to the differentiation or specification of different types of adaptive immune T cells. An antigen is phagocytosed and processed by innate immune cells, which then migrate to the nearest lymph node and present the antigenic peptide to naïve T cells. Depending on the type of antigen, costimulatory molecules on the innate immune cell, and the combinations of specific cytokines, naïve T cells mature into specific subtypes that best serve the inflammatory effort to clear the pathogenic stimulus. CD4 T cells, or T helper (Th) cells, can be divided into four principle groups, Th1, Th2, Th17, and T regulatory cells (Treg). Th1 cells are mainly involved in regulating immune responses to intracellular microorganisms and organ-specific autoimmune diseases; Th2 are critical for immunity against extracellular pathogens, such as helminths, and are responsible for allergic inflammatory diseases; Th17 cells play a central role in protection against microbial challenges, such as extracellular bacteria and fungi; Treg cells are involved in maintaining self tolerance and regulating immune responses. This T cell maturation process appears to be heavily influenced by sensory neuronal mediators. Neuropeptides, such as CGRP and VIP, can bias dendritic cells towards a Th2-type immunity and reduce Th1-type immunity by promoting the production of certain cytokines and inhibiting others, as well as by reducing or enhancing dendritic cell migration to local lymph nodes 8, 10, 43. Sensory neurons also contribute considerably to allergic (mainly Th2 driven) inflammation 17. In addition to regulating Th1 and Th2 cells, other neuropeptides, such as SP and Hemokinin-1, can drive the inflammatory response more toward Th17 or Treg 44, 45, which means that neurons may also be involved in regulating inflammatory resolution. In immunopathologies such as colitis and psoriasis, blockade of neuronal mediators like substance P may significantly dampen T cell and immune mediated damage 15–17, although antagonizing one mediator may by itself only have a limited effect on neurogenic inflammation.
Considering that signaling molecules released from peripheral sensory nerve fibers regulate not only small blood vessels, but also the chemotaxis, homing, maturation, and activation of immune cells, it is becoming clear that neuro-immune interactions are much more intricate than previously thought (Fig. 2). Furthermore, it is quite conceivable that it is not individual neural mediators but rather specific combinations of signaling molecules released from nociceptors that influence different stages and types of immune responses.
Autonomic Reflex Control of Immunity
A role for a cholinergic autonomic nervous system “reflex” circuit in the regulation of peripheral immune responses also appears prominent 46. The vagus is the chief parasympathetic nerve connecting the brainstem with visceral organs. Work by Kevin Tracey and others point to potent generalized anti-inflammatory responses in septic shock and endotoxemia, triggered by an efferent vagal nerve activity leading to a suppression of peripheral macrophages 47–49. The vagus activates peripheral adrenergic celiac ganglion neurons innervating the spleen, leading to the downstream release of acetylcholine, which binds to alpha-7 nicotinic receptors on macrophages in the spleen and gastrointestinal tract. This induces activation of the JAK2/STAT3 SOCS3 signaling pathway, which powerfully suppresses TNF-alpha transcription 47. The adrenergic celiac ganglion also directly communicates with a subset of acetylcholine producing memory T cells, which suppress inflammatory macrophages 48.
Invariant natural Killer T cells (iNKT) are a specialized subset of T cells that recognize microbial lipids in the context of CD1d instead of peptide antigens. NKT cells are a key lymphocyte population involved in the combat of infectious pathogens and regulation of systemic immunity. NKT cells reside and traffic mainly through the vasculature and sinusoids of the spleen and liver. Sympathetic beta-adrenergic nerves in the liver directly signal to modulate NKT cell activity 50. During a mouse model of stroke (MCAO), for example, liver NKT cell mobility was visibly suppressed, which was reversed by sympathetic denervation or beta-adrenergic antagonists. Furthermore, this immunosuppressive activity of noradrenergic neurons on NKT cells led to increases in systemic infection and lung injury. Therefore, efferent signals from autonomic neurons can mediate a potent immuno-suppression.

Dr. Alex Jimenez’s Insight
Neurogenic inflammation is a local inflammatory response generated by the nervous system. It is believed to play a fundamental role in the pathogenesis of a variety of health issues, including, migraine, psoriasis, asthma, fibromyalgia, eczema, rosacea, dystonia and multiple chemical sensitivity. Although neurogenic inflammation associated with the peripheral nervous system has been extensively researched, the concept of neurogenic inflammation within the central nervous system still needs further research. According to several research studies, however, magnesium deficiencies are believed to be the main cause for neurogenic inflammation. The following article demonstrates an overview of the mechanisms of neurogenic inflammation in the nervous system, which may help healthcare professionals determine the best treatment approach to care for a variety of health issues associated with the nervous system.
Conclusions
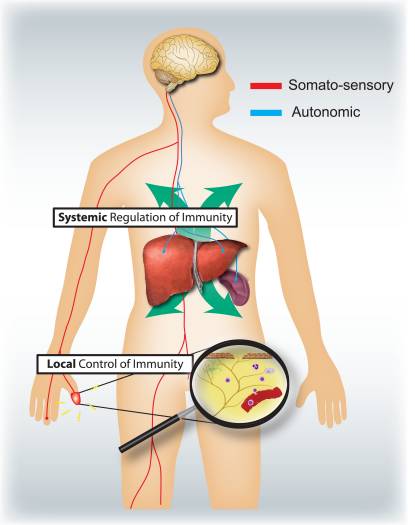
What are the respective specific roles of the somatosensory and autonomic nervous systems in regulating inflammation and the immune system (Fig. 4)? Activation of nociceptors leads to local axon reflexes, which locally recruit and activate immune cells and is therefore, mainly pro-inflammatory and spatially confined. In contrast, autonomic stimulation leads to a systemic immunosuppression by affecting pools of immune cells in liver and spleen. The afferent signaling mechanisms in the periphery leading to the triggering of the immunosuppressive vagal cholinergic reflex circuit are poorly understood. However, 80–90% of vagal fibers are primary afferent sensory fibers, and therefore signals from the viscera, many potentially driven by immune cells, may lead to activation of interneurons in the brainstem and through them to an output in efferent vagal fibers 46.
Typically, the time course and nature of inflammation, whether during infection, allergic reactions, or auto-immune pathologies, is defined by the categories of immune cells involved. It will be important to know what different types of immune cells are regulated by sensory and autonomic signals. A systematic assessment of what mediators can be released from nociceptors and autonomic neurons and the expression of receptors for these by different innate and adaptive immune cells might help address this question.
During evolution, similar danger detection molecular pathways have developed for both innate immunity and nociception even though the cells have completely different developmental lineages. While PRRs and noxious ligand-gated ion channels are studied separately by immunologists and neurobiologists, the line between these two fields is increasingly blurred. During tissue damage and pathogenic infection, release of danger signals are likely to lead to a coordinated activation of both peripheral neurons and immune cells with complex bidirectional communication, and an integrated host defense. The anatomical positioning of nociceptors at the interface with the environment, the speed of neural transduction and their ability to release potent cocktails of immune-acting mediators allows the peripheral nervous system to actively modulate the innate immune response and coordinate downstream adaptive immunity. Conversely, nociceptors are highly sensitive to immune mediators, which activate and sensitize the neurons. Neurogenic and immune-mediated inflammation are not, therefore, independent entities but act together as early warning devices. However, the peripheral nervous system also plays an important role in the pathophysiology, and perhaps etiology, of many immune diseases like asthma, psoriasis, or colitis because its capacity to activate the immune system can amplify pathological inflammation 15–17. Treatment for immune disorders may need to include, therefore, the targeting of nociceptors as well as of immune cells.
Acknowledgements
We thank the NIH for support (2R37NS039518).
In conclusion, understanding the role of neurogenic inflammation when it comes to host defense and immunopathology is essential towards determining the proper treatment approach for a variety of nervous system health issues. By looking at the interactions of the peripheral neurons with immune cells, healthcare professionals may advance therapeutic approaches to further help increase host defense as well as suppress immunopathology. The purpose of the article above is to help patients understand the clinical neurophysiology of neuropathy, among other nerve injury health issues. Information referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic as well as to spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
1. Sauer SK, Reeh PW, Bove GM. Noxious heat-induced CGRP release from rat sciatic nerve axons in vitro. Eur J Neurosci. 2001;14:1203–1208. [PubMed]
2. Edvinsson L, Ekman R, Jansen I, McCulloch J, Uddman R. Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab. 1987;7:720–728. [PubMed]
3. McCormack DG, Mak JC, Coupe MO, Barnes PJ. Calcitonin gene-related peptide vasodilation of human pulmonary vessels. J Appl Physiol. 1989;67:1265–1270. [PubMed]
4. Saria A. Substance P in sensory nerve fibres contributes to the development of oedema in the rat hind paw after thermal injury. Br J Pharmacol. 1984;82:217–222. [PMC free article] [PubMed]
5. Brain SD, Williams TJ. Interactions between the tachykinins and calcitonin generelated peptide lead to the modulation of oedema formation and blood flow in rat skin. Br J Pharmacol. 1989;97:77–82. [PMC free article] [PubMed]
6. Fryer AD, et al. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J Clin Invest. 2006;116:228–236. [PMC free article] [PubMed]
7. Ansel JC, Brown JR, Payan DG, Brown MA. Substance P selectively activates TNF-alpha gene expression in murine mast cells. J Immunol. 1993;150:4478–4485. [PubMed]
8. Ding W, Stohl LL, Wagner JA, Granstein RD. Calcitonin gene-related peptide biases Langerhans cells toward Th2-type immunity. J Immunol. 2008;181:6020–6026. [PMC free article] [PubMed]
9. Hosoi J, et al. Regulation of Langerhans cell function by nerves containing calcitonin gene-related peptide. Nature. 1993;363:159–163. [PubMed]
10. Mikami N, et al. Calcitonin gene-related peptide is an important regulator of cutaneous immunity: effect on dendritic cell and T cell functions. J Immunol. 2011;186:6886–6893. [PubMed]
11. Rochlitzer S, et al. The neuropeptide calcitonin gene-related peptide affects allergic airway inflammation by modulating dendritic cell function. Clin Exp Allergy. 2011;41:1609–1621. [PubMed]
12. Cyphert JM, et al. Cooperation between mast cells and neurons is essential for antigen-mediated bronchoconstriction. J Immunol. 2009;182:7430–7439. [PMC free article] [PubMed]
13. Levine JD, et al. Intraneuronal substance P contributes to the severity of experimental arthritis. Science. 1984;226:547–549. [PubMed]
14. Levine JD, Khasar SG, Green PG. Neurogenic inflammation and arthritis. Ann N Y Acad Sci. 2006;1069:155–167. [PubMed]
15. Engel MA, et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology. 2011;141:1346–1358. [PubMed]
16. Ostrowski SM, Belkadi A, Loyd CM, Diaconu D, Ward NL. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530–1538. [PMC free article] [PubMed]
17. Caceres AI, et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc Natl Acad Sci U S A. 2009;106:9099–9104. [PMC free article] [PubMed]
18. Caterina MJ, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. [PubMed]
19. Bessac BF, et al. Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases. FASEB J. 2009;23:1102–1114. [PMC free article] [PubMed]
20. Cruz-Orengo L, et al. Cutaneous nociception evoked by 15-delta PGJ2 via activation of ion channel TRPA1. Mol Pain. 2008;4:30. [PMC free article] [PubMed]
21. Trevisani M, et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A. 2007;104:13519–13524. [PMC free article] [PubMed]
22. Janeway CA, Jr, Medzhitov R. Introduction: the role of innate immunity in the adaptive immune response. Semin Immunol. 1998;10:349–350. [PubMed]
23. Matzinger P. An innate sense of danger. Ann N Y Acad Sci. 2002;961:341–342. [PubMed]
24. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. [PubMed]
25. Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus. Nat Neurosci. 2010;13:1460–1462. [PMC free article] [PubMed]
26. Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res. 2011;90:759–764. [PubMed]
27. Qi J, et al. Painful pathways induced by TLR stimulation of dorsal root ganglion neurons. J Immunol. 2011;186:6417–6426. [PMC free article] [PubMed]
28. Cockayne DA, et al. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015. [PubMed]
29. Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. [PubMed]
30. Souslova V, et al. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–1017. [PubMed]
31. de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. [PubMed]
32. Link TM, et al. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat Immunol. 2010;11:232–239. [PMC free article] [PubMed]
33. Turner H, del Carmen KA, Stokes A. Link between TRPV channels and mast cell function. Handb Exp Pharmacol. 2007:457–471. [PubMed]
34. Binshtok AM, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062–14073. [PMC free article] [PubMed]
35. Zhang XC, Kainz V, Burstein R, Levy D. Tumor necrosis factor-alpha induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain. 2011;152:140–149. [PMC free article] [PubMed]
36. Samad TA, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. [PubMed]
37. Veres TZ, et al. Spatial interactions between dendritic cells and sensory nerves in allergic airway inflammation. Am J Respir Cell Mol Biol. 2007;37:553–561. [PubMed]
38. Smith CH, Barker JN, Morris RW, MacDonald DM, Lee TH. Neuropeptides induce rapid expression of endothelial cell adhesion molecules and elicit granulocytic infiltration in human skin. J Immunol. 1993;151:3274–3282. [PubMed]
39. Dunzendorfer S, Meierhofer C, Wiedermann CJ. Signaling in neuropeptide-induced migration of human eosinophils. J Leukoc Biol. 1998;64:828–834. [PubMed]
40. Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M. Human T cells express a functional ionotropic glutamate receptor GluR3, and glutamate by itself triggers integrin-mediated adhesion to laminin and fibronectin and chemotactic migration. J Immunol. 2003;170:4362–4372. [PubMed]
41. Czepielewski RS, et al. Gastrin-releasing peptide receptor (GRPR) mediates chemotaxis in neutrophils. Proc Natl Acad Sci U S A. 2011;109:547–552. [PMC free article] [PubMed]
42. Brogden KA, Guthmiller JM, Salzet M, Zasloff M. The nervous system and innate immunity: the neuropeptide connection. Nat Immunol. 2005;6:558–564. [PubMed]
43. Jimeno R, et al. Effect of VIP on the balance between cytokines and master regulators of activated helper T cells. Immunol Cell Biol. 2011;90:178–186. [PubMed]
44. Razavi R, et al. TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell. 2006;127:1123–1135. [PubMed]
45. Cunin P, et al. The tachykinins substance P and hemokinin-1 favor the generation of human memory Th17 cells by inducing IL-1beta, IL-23, and TNF-like 1A expression by monocytes. J Immunol. 2011;186:4175–4182. [PubMed]
46. Andersson U, Tracey KJ. Reflex Principles of Immunological Homeostasis. Annu Rev Immunol. 2011 [PMC free article] [PubMed]
47. de Jonge WJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–851. [PubMed]
48. Rosas-Ballina M, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. [PMC free article] [PubMed]
49. Wang H, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. [PubMed]
50. Wong CH, Jenne CN, Lee WY, Leger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. 2011;334:101–105. [PubMed]

Additional Topics: Back Pain
Back pain is one of the most prevalent causes for disability and missed days at work worldwide. As a matter of fact, back pain has been attributed as the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience some type of back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.

EXTRA IMPORTANT TOPIC: Back Pain Management
MORE TOPICS: EXTRA EXTRA: El Paso, TX | Chronic Pain Treatment
Post Disclaimer
General Disclaimer, Licenses and Board Certifications *
Professional Scope of Practice *
The information herein on "The Role of Neurogenic Inflammation" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a Multi-State board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our multidisciplinary team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those on this site and on our family practice-based chiromed.com site, focusing on naturally restoring health for patients of all ages.
Our areas of multidisciplinary practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is multidisciplinary, focusing on musculoskeletal and physical medicine; wellness; contributing etiological viscerosomatic disturbances within clinical presentations; associated somato-visceral reflex clinical dynamics; subluxation complexes; sensitive health issues; and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and licensure jurisdiction. We use functional health & wellness protocols to treat and support care for musculoskeletal injuries or disorders.
Our videos, posts, topics, and insights address clinical matters and issues that directly or indirectly relate to our clinical scope of practice.
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies upon request to regulatory boards and the public.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Multidisciplinary Licensing & Board Certifications:
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License #: TX5807, Verified: TX5807
New Mexico DC License #: NM-DC2182, Verified: NM-DC2182
Multi-State Advanced Practice Registered Nurse (APRN*) in Texas & Multi-States
Multi-state Compact APRN License by Endorsement (42 States)
Texas APRN License #: 1191402, Verified: 1191402 *
New Mexico CNP License#: 90560, Verified
Florida APRN License #: 11043890, Verified: APRN11043890 *
Colorado License #: C-APN.0105610-C-NP, Verified: C-APN.0105610-C-NP
New York License #: N25929, Verified N25929
License Verification Link: Nursys License Verifier
* Prescriptive Authority Authorized
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
| Yes | 363LF0000X - Nurse Practitioner - Family | NM |
90560 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
📆 Schedule Appointment: Schedule 24/7 (Click Here)