


Epigenetic Abstract:
The increased prevalence of obesity and related comorbidities is a major public health problem. While genetic factors undoubtedly play a role in determining individual susceptibility to weight gain and obesity, the identified genetic variants only explain part of the variation. This has led to growing interest in understanding the potential role of epigenetics as a mediator of gene-environment interactions underlying the development of obesity and its associated comorbidities. Initial evidence in support of a role of epigenetics in obesity and type 2 diabetes mellitus (T2DM) was mainly provided by animal studies, which reported epigenetic changes in key metabolically important tissues following high-fat feeding and epigenetic differences between lean and obese animals and by human studies which showed epigenetic changes in obesity and T2DM candidate genes in obese/diabetic individuals. More recently, advances in epigenetic methodologies and the reduced cost of epigenome-wide association studies (EWAS) have led to a rapid expansion of studies in human populations. These studies have also reported epigenetic differences between obese/T2DM adults and healthy controls and epigenetic changes in association with nutritional, weight loss, and exercise interventions. There is also increasing evidence from both human and animal studies that the relationship between perinatal nutritional exposures and later risk of obesity and T2DM may be mediated by epigenetic changes in the offspring. The aim of this review is to summarize the most recent developments in this rapidly moving field, with a particular focus on human EWAS and studies investigating the impact of nutritional and lifestyle factors (both pre- and postnatal) on the epigenome and their relationship to metabolic health outcomes. The difficulties in distinguishing consequence from causality in these studies and the critical role of animal models for testing causal relationships and providing insight into underlying mechanisms are also addressed. In summary, the area of epigenetics and metabolic health has seen rapid developments in a short space of time. While the outcomes to date are promising, studies are ongoing, and the next decade promises to be a time of productive research into the complex interactions between the genome, epigenome, and environment as they relate to metabolic disease.
Keywords: Epigenetics, DNA methylation, Obesity, Type 2 diabetes, Developmental programming
Table of Contents
Introduction
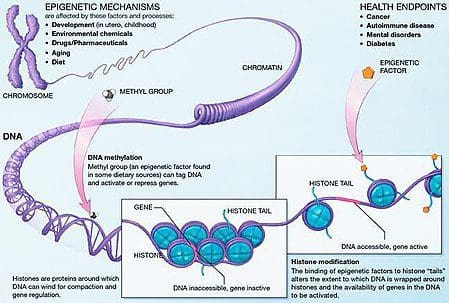 Obesity is a complex, multifactorial disease, and better understanding of the mechanisms underlying the interactions between lifestyle, environment, and genetics is critical for developing effective strategies for prevention and treatment [1].
Obesity is a complex, multifactorial disease, and better understanding of the mechanisms underlying the interactions between lifestyle, environment, and genetics is critical for developing effective strategies for prevention and treatment [1].
In a society where energy-dense food is plentiful and the need for physical activity is low, there is a wide variation in individuals’ susceptibility to develop obesity and metabolic health problems. Estimates of the role of heredity in this variation are in the range of 40–70 %, and while large genome-wide association studies (GWAS) have identified a number of genetic loci associated with obesity risk, the ~100 most common genetic variants only account for a few percent of variance in obesity [2, 3]. Genome-wide estimates are higher, accounting for ~20 % of the variation [3]; however, a large portion of the heritability remains unexplained.
Recently, attention has turned to investigating the role of epigenetic changes in the etiology of obesity. It has been argued that the epigenome may represent the mechanistic link between genetic variants and environmental factors in determining obesity risk and could help explain the “missing heritability.” The first human epigenetic studies were small and only investigated a limited number of loci. While this generally resulted in poor reproducibility, some of these early findings, for instance the relationship between PGC1A methylation and type 2 diabetes mellitus (T2DM) [4] and others as discussed in van Dijk et al. [5], have been replicated in later studies. Recent advances and increased affordability of high- throughput technologies now allow for large-scale epigenome wide association studies (EWAS) and integration of different layers of genomic information to explore the complex interactions between the genotype, epigenome, transcriptome, and the environment [6–9]. These studies are still in their infancy, but the results thus far have shown promise in helping to explain the variation in obesity susceptibility.
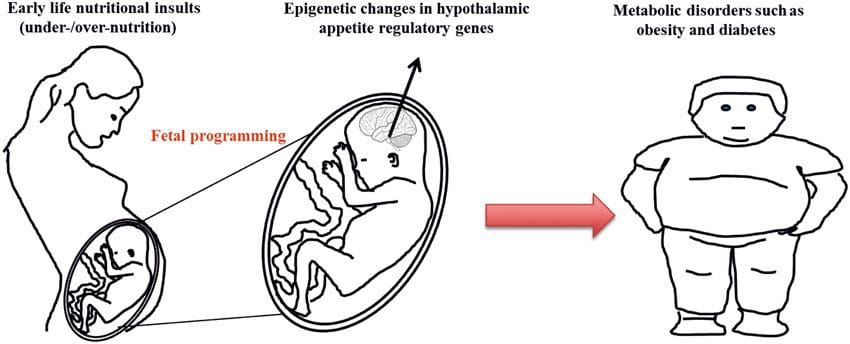
There is increasing evidence that obesity has develop mental origins, as exposure to a suboptimal nutrient supply before birth or in early infancy is associated with an increased risk of obesity and metabolic disease in later life [10–13]. Initially, animal studies demonstrated that a range of early life nutritional exposures, especially those experienced early in gestation, could induce epigenetic changes in key metabolic tissues of the offspring that persisted after birth and result in permanent alterations in gene function [13–17]. Evidence is emerging to support the existence of the same mechanism in humans. This has led to a search for epigenetic marks present early in life that predict later risk of metabolic disease, and studies to determine whether epigenetic programming of metabolic disease could be prevented or reversed in later life.
This review provides an update of our previous systematic review of studies on epigenetics and obesity in humans [5]. Our previous review showcased the promising outcomes of initial studies, including the first potential epigenetic marks for obesity that could be detected at birth (e.g., RXRA) [18]. However, it also highlighted the limited reproducibility of the findings and the lack of larger scale longitudinal investigations. The current review focuses on recent developments in this rapidly moving field and, in particular, on human EWAS and studies investigating the impact of (pre- and postnatal) nutritional and lifestyle factors on the epigenome and the emerging role of epigenetics in the pathology of obesity. We also address the difficulties in identifying causality in these studies and the importance of animal models in providing insight into mechanisms.
Review
Epigenetic Changes In Animal Models Of Obesity
 Animal models provide unique opportunities for highly controlled studies that provide mechanistic insight into the role of specific epigenetic marks, both as indicators of current metabolic status and as predictors of the future risk of obesity and metabolic disease. A particularly important aspect of animal studies is that they allow for the assessment of epigenetic changes within target tissues, including the liver and hypothalamus, which is much more difficult in humans. Moreover, the ability to harvest large quantities of fresh tissue makes it possible to assess multiple chromatin marks as well as DNA methylation. Some of these epigenetic modifications either alone or in combination may be responsive to environmental programming. In animal models, it is also possible to study multiple generations of offspring and thus enable differentiation between trans-generational and intergenerational transmission of obesity risk mediated by epigenetic memory of parental nutritional status, which cannot be easily distinguished in human studies. We use the former term for meiotic transmission of risk in the absence of continued exposure while the latter primarily entails direct transmission of risk through metabolic reprogramming of the fetus or gametes.
Animal models provide unique opportunities for highly controlled studies that provide mechanistic insight into the role of specific epigenetic marks, both as indicators of current metabolic status and as predictors of the future risk of obesity and metabolic disease. A particularly important aspect of animal studies is that they allow for the assessment of epigenetic changes within target tissues, including the liver and hypothalamus, which is much more difficult in humans. Moreover, the ability to harvest large quantities of fresh tissue makes it possible to assess multiple chromatin marks as well as DNA methylation. Some of these epigenetic modifications either alone or in combination may be responsive to environmental programming. In animal models, it is also possible to study multiple generations of offspring and thus enable differentiation between trans-generational and intergenerational transmission of obesity risk mediated by epigenetic memory of parental nutritional status, which cannot be easily distinguished in human studies. We use the former term for meiotic transmission of risk in the absence of continued exposure while the latter primarily entails direct transmission of risk through metabolic reprogramming of the fetus or gametes.
Animal studies have played a critical role in our current understanding of the role of epigenetics in the developmental origins of obesity and T2DM. Both increased and decreased maternal nutrition during pregnancy have been associated with increased fat deposition in offspring of most mammalian species studied to date (reviewed in [11, 13–15, 19]). Maternal nutrition during pregnancy not only has potential for direct effects on the fetus, it also may directly impact the developing oocytes of female fetuses and primordial germ cells of male fetuses and therefore could impact both the off- spring and grand-offspring. Hence, multigenerational data are usually required to differentiate between maternal intergenerational and trans-generational transmission mechanisms.
Table 1 summarizes a variety of animal models that have been used to provide evidence of metabolic and epigenetic changes in offspring associated with the parental plane of nutrition. It also contains information pertaining to studies identifying altered epigenetic marks in adult individuals who undergo direct nutritional challenges. The table is structured by suggested risk transmission type.
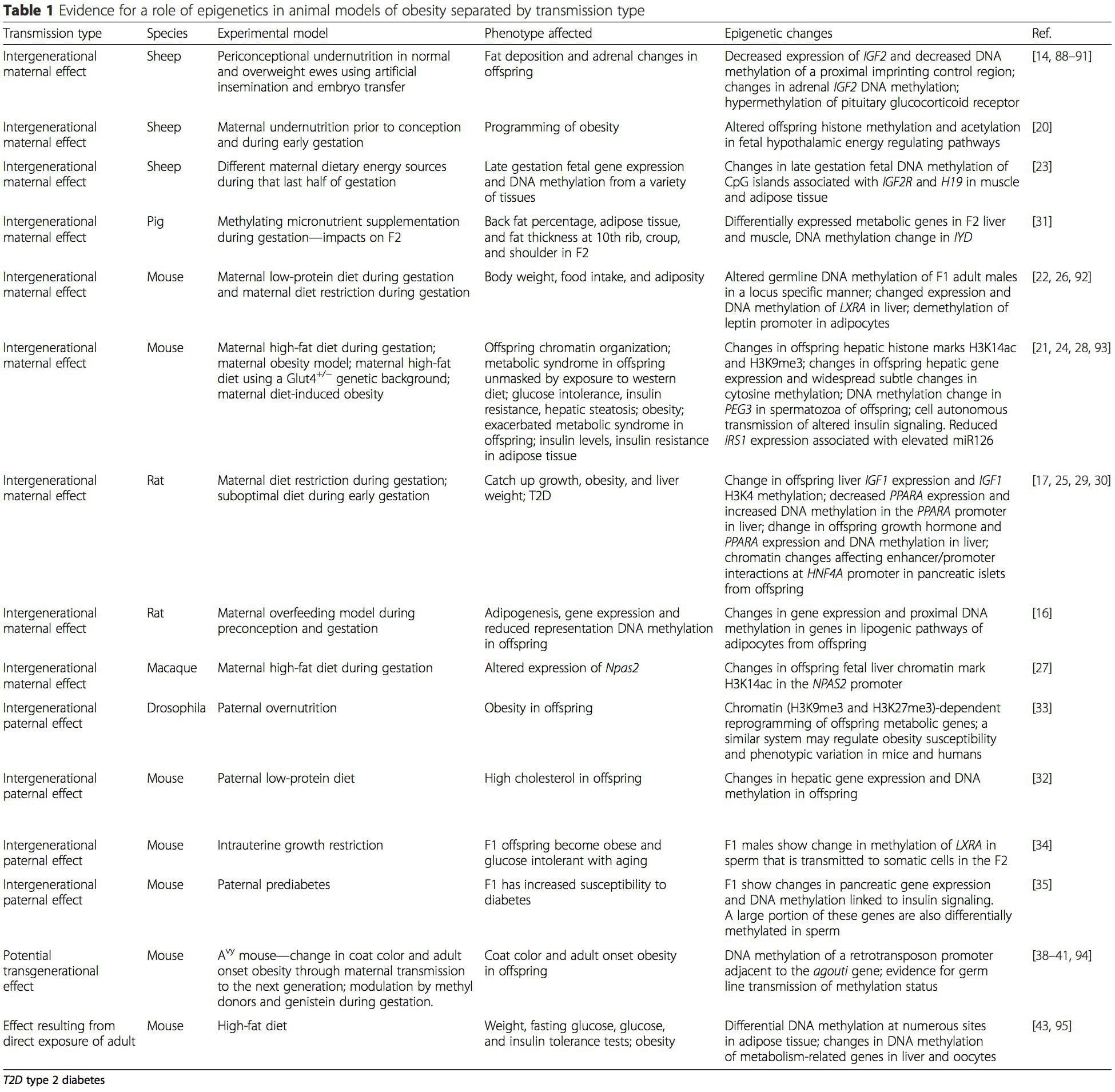 (i) Epigenetic Changes In Offspring Associated With Maternal Nutrition During Gestation
(i) Epigenetic Changes In Offspring Associated With Maternal Nutrition During Gestation
 Maternal nutritional supplementation, undernutrition, and over nutrition during pregnancy can alter fat deposition and energy homeostasis in offspring [11, 13–15, 19]. Associated with these effects in the offspring are changes in DNA methylation, histone post-translational modifications, and gene expression for several target genes, especially genes regulating fatty acid metabolism and insulin signaling [16, 17, 20–30]. The diversity of animal models used in these studies and the common metabolic pathways impacted suggest an evolutionarily conserved adaptive response mediated by epigenetic modification. However, few of the specific identified genes and epigenetic changes have been cross-validated in related studies, and large-scale genome-wide investigations have typically not been applied. A major hindrance to comparison of these studies is the different develop mental windows subjected to nutritional challenge, which may cause considerably different outcomes. Proof that the epigenetic changes are causal rather than being associated with offspring phenotypic changes is also required. This will necessitate the identification of a parental nutritionally induced epigenetic “memory” response that precedes development of the altered phenotype in offspring.
Maternal nutritional supplementation, undernutrition, and over nutrition during pregnancy can alter fat deposition and energy homeostasis in offspring [11, 13–15, 19]. Associated with these effects in the offspring are changes in DNA methylation, histone post-translational modifications, and gene expression for several target genes, especially genes regulating fatty acid metabolism and insulin signaling [16, 17, 20–30]. The diversity of animal models used in these studies and the common metabolic pathways impacted suggest an evolutionarily conserved adaptive response mediated by epigenetic modification. However, few of the specific identified genes and epigenetic changes have been cross-validated in related studies, and large-scale genome-wide investigations have typically not been applied. A major hindrance to comparison of these studies is the different develop mental windows subjected to nutritional challenge, which may cause considerably different outcomes. Proof that the epigenetic changes are causal rather than being associated with offspring phenotypic changes is also required. This will necessitate the identification of a parental nutritionally induced epigenetic “memory” response that precedes development of the altered phenotype in offspring.
(ii)Effects Of Paternal Nutrition On Offspring Epigenetic Marks
 Emerging studies have demonstrated that paternal plane of nutrition can impact offspring fat deposition and epigenetic marks [31–34]. One recent investigation using mice has demonstrated that paternal pre-diabetes leads to increased susceptibility to diabetes in F1 offspring with associated changes in pancreatic gene expression and DNA methylation linked to insulin signaling [35]. Importantly, there was an overlap of these epigenetic changes in pancreatic islets and sperm suggesting germ line inheritance. However, most of these studies, although intriguing in their implications, are limited in the genomic scale of investigation and frequently show weak and somewhat transient epigenetic alterations associated with mild metabolic phenotypes in offspring.
Emerging studies have demonstrated that paternal plane of nutrition can impact offspring fat deposition and epigenetic marks [31–34]. One recent investigation using mice has demonstrated that paternal pre-diabetes leads to increased susceptibility to diabetes in F1 offspring with associated changes in pancreatic gene expression and DNA methylation linked to insulin signaling [35]. Importantly, there was an overlap of these epigenetic changes in pancreatic islets and sperm suggesting germ line inheritance. However, most of these studies, although intriguing in their implications, are limited in the genomic scale of investigation and frequently show weak and somewhat transient epigenetic alterations associated with mild metabolic phenotypes in offspring.
(iii)Potential Trans-generational Epigenetic Changes Promoting Fat Deposition In Offspring
 Stable transmission of epigenetic information across multiple generations is well described in plant systems and C. elegans, but its significance in mammals is still much debated [36, 37]. An epigenetic basis for grand- parental transmission of phenotypes in response to dietary exposures has been well established, including in livestock species [31]. The most influential studies demonstrating effects of epigenetic transmission impacting offspring phenotype have used the example of the viable yellow agouti (Avy) mouse [38]. In this mouse, an insertion of a retrotransposon upstream of the agouti gene causes its constitutive expression and consequent yellow coat color and adult onset obesity. Maternal transmission through the germ line results in DNA methylation mediated silencing of agouti expression resulting in wild-type coat color and lean phenotype of the offspring [39, 40]. Importantly, subsequent studies in these mice demonstrated that maternal exposure to methyl donors causes a shift in coat color [41]. One study has reported transmission of a phenotype to the F3 generation and alterations in expression of large number of genes in response to protein restriction in F0 [42]; however, alterations in expression were highly variable and a direct link to epigenetic changes was not identified in this system.
Stable transmission of epigenetic information across multiple generations is well described in plant systems and C. elegans, but its significance in mammals is still much debated [36, 37]. An epigenetic basis for grand- parental transmission of phenotypes in response to dietary exposures has been well established, including in livestock species [31]. The most influential studies demonstrating effects of epigenetic transmission impacting offspring phenotype have used the example of the viable yellow agouti (Avy) mouse [38]. In this mouse, an insertion of a retrotransposon upstream of the agouti gene causes its constitutive expression and consequent yellow coat color and adult onset obesity. Maternal transmission through the germ line results in DNA methylation mediated silencing of agouti expression resulting in wild-type coat color and lean phenotype of the offspring [39, 40]. Importantly, subsequent studies in these mice demonstrated that maternal exposure to methyl donors causes a shift in coat color [41]. One study has reported transmission of a phenotype to the F3 generation and alterations in expression of large number of genes in response to protein restriction in F0 [42]; however, alterations in expression were highly variable and a direct link to epigenetic changes was not identified in this system.
(iv) Direct Exposure Of Individuals To Excess Nutrition In Postnatal Life
 While many studies have identified diet-associated epigenetic changes in animal models using candidate site-specific regions, there have been few genome-wide analyses undertaken. A recent study focussed on determining the direct epigenetic impact of high-fat diets/ diet-induced obesity in adult mice using genome-wide gene expression and DNA methylation analyses [43]. This study identified 232 differentially methylated regions (DMRs) in adipocytes from control and high-fat fed mice. Importantly, the corresponding human regions for the murine DMRs were also differentially methylated in adipose tissue from a population of obese and lean humans, thereby highlighting the remarkable evolutionary conservation of these regions. This result emphasizes the likely importance of the identified DMRs in regulating energy homeostasis in mammals.
While many studies have identified diet-associated epigenetic changes in animal models using candidate site-specific regions, there have been few genome-wide analyses undertaken. A recent study focussed on determining the direct epigenetic impact of high-fat diets/ diet-induced obesity in adult mice using genome-wide gene expression and DNA methylation analyses [43]. This study identified 232 differentially methylated regions (DMRs) in adipocytes from control and high-fat fed mice. Importantly, the corresponding human regions for the murine DMRs were also differentially methylated in adipose tissue from a population of obese and lean humans, thereby highlighting the remarkable evolutionary conservation of these regions. This result emphasizes the likely importance of the identified DMRs in regulating energy homeostasis in mammals.
Human Studies
Drawing on the evidence from animal studies and with the increasing availability of affordable tools for genome- wide analysis, there has been a rapid expansion of epigenome studies in humans. These studies have mostly focused on the identification of site-specific differences in DNA methylation that are associated with metabolic phenotypes.
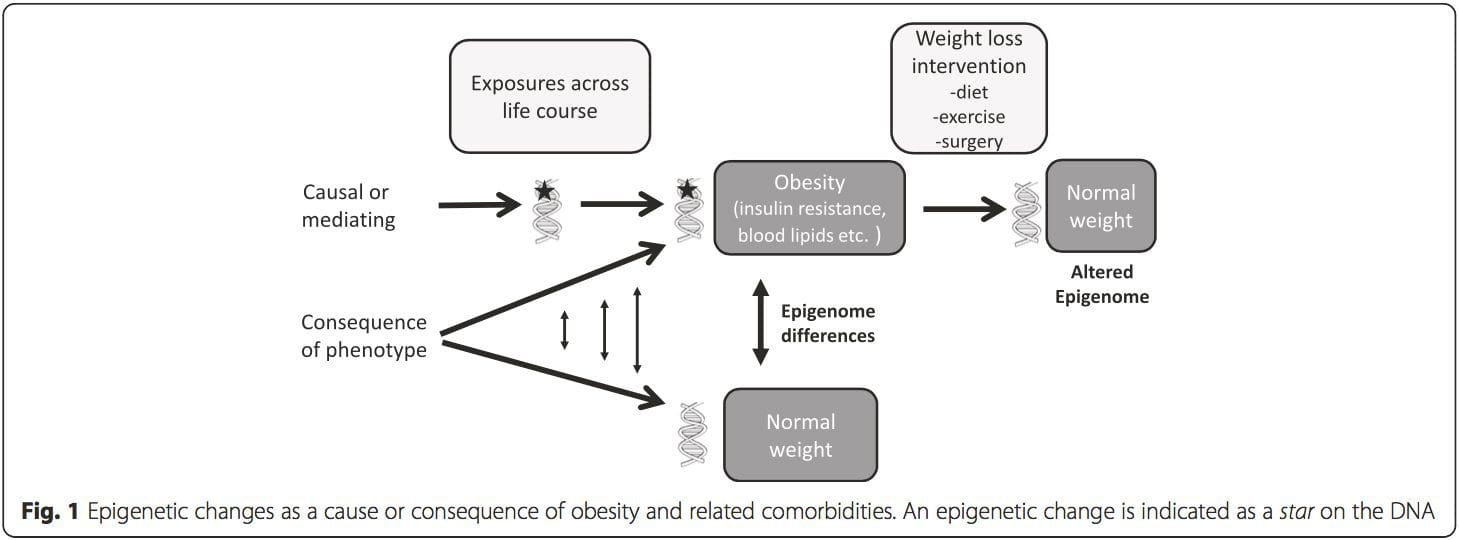
A key question is the extent to which epigenetic modifications contribute to the development of the metabolic phenotype, rather than simply being a con- sequence of it (Fig. 1). Epigenetic programming could contribute to obesity development, as well as playing a role in consequent risk of cardiovascular and metabolic problems. In human studies, it is difficult to prove causality [44], but inferences can be made from a number of lines of evidence:
 (i) Genetic association studies. Genetic polymorphisms that are associated with an increased risk of developing particular conditions are a priori linked to the causative genes. The presence of differential methylation in such regions infers functional relevance of these epigenetic changes in controlling expression of the proximal gene(s). There are strong cis-acting genetic effects underpinning much epigenetic variation [7, 45], and in population-based studies, methods that use genetic surrogates to infer a causal or mediating role of epigenome differences have been applied [7, 46–48]. The use of familial genetic information can also lead to the identification of potentially causative candidate regions showing phenotype-related differential methylation [49].
(i) Genetic association studies. Genetic polymorphisms that are associated with an increased risk of developing particular conditions are a priori linked to the causative genes. The presence of differential methylation in such regions infers functional relevance of these epigenetic changes in controlling expression of the proximal gene(s). There are strong cis-acting genetic effects underpinning much epigenetic variation [7, 45], and in population-based studies, methods that use genetic surrogates to infer a causal or mediating role of epigenome differences have been applied [7, 46–48]. The use of familial genetic information can also lead to the identification of potentially causative candidate regions showing phenotype-related differential methylation [49].
(ii)Timing of epigenetic changes. The presence of an epigenetic mark prior to development of a phenotype is an essential feature associated with causality. Conversely, the presence of a mark in association with obesity, but not before its development, can be used to exclude causality but would not exclude a possible role in subsequent obesity-related pathology.
(iii)Plausible inference of mechanism. This refers to epigenetic changes that are associated with altered expression of genes with an established role in regulating the phenotype of interest. One such example is the association of methylation at two CpG sites at the CPT1A gene with circulating triglyceride levels [50]. CPT1A encodes carnitine palmitoyltransferase 1A, an enzyme with a central role in fatty acid metabolism, and this is strongly indicative that differential methylation of this gene may be causally related to the alterations in plasma triglyceride concentrations.
Epigenome-Wide Association Studies: Identifying Epigenetic Biomarkers Of Metabolic Health
A number of recent investigations have focused on exploring associations between obesity/metabolic diseases and DNA methylation across the genome (Table 2). The largest published EWAS so far, including a total of 5465 individuals, identified 37 methylation sites in blood that were associated with body mass index (BMI), including sites in CPT1A, ABCG1, and SREBF1 [51]. Another large-scale study showed consistent associations between BMI and methylation in HIF3A in whole blood and adipose tissue [52], a finding which was also partially replicated in other studies [9, 51]. Other recently reported associations between obesity-related measures and DNA methylation include (i) DNA methylation differences between lean and obese individuals in LY86 in blood leukocytes [53]; (ii) associations between PGC1A promoter methylation in whole blood of children and adiposity 5 years later [54]; (iii) associations between waist-hip ratio and ADRB3 methylation in blood [55]; and (iv) associations between BMI, body fat distribution measures, and multiple DNA methylation sites in adipose tissue [9, 56]. EWAS have also shown associations between DNA methylation sites and blood lipids [55, 57–59], serum metabolites [60], insulin resistance [9, 61], and T2DM [48, 62, 63] (Table 2).
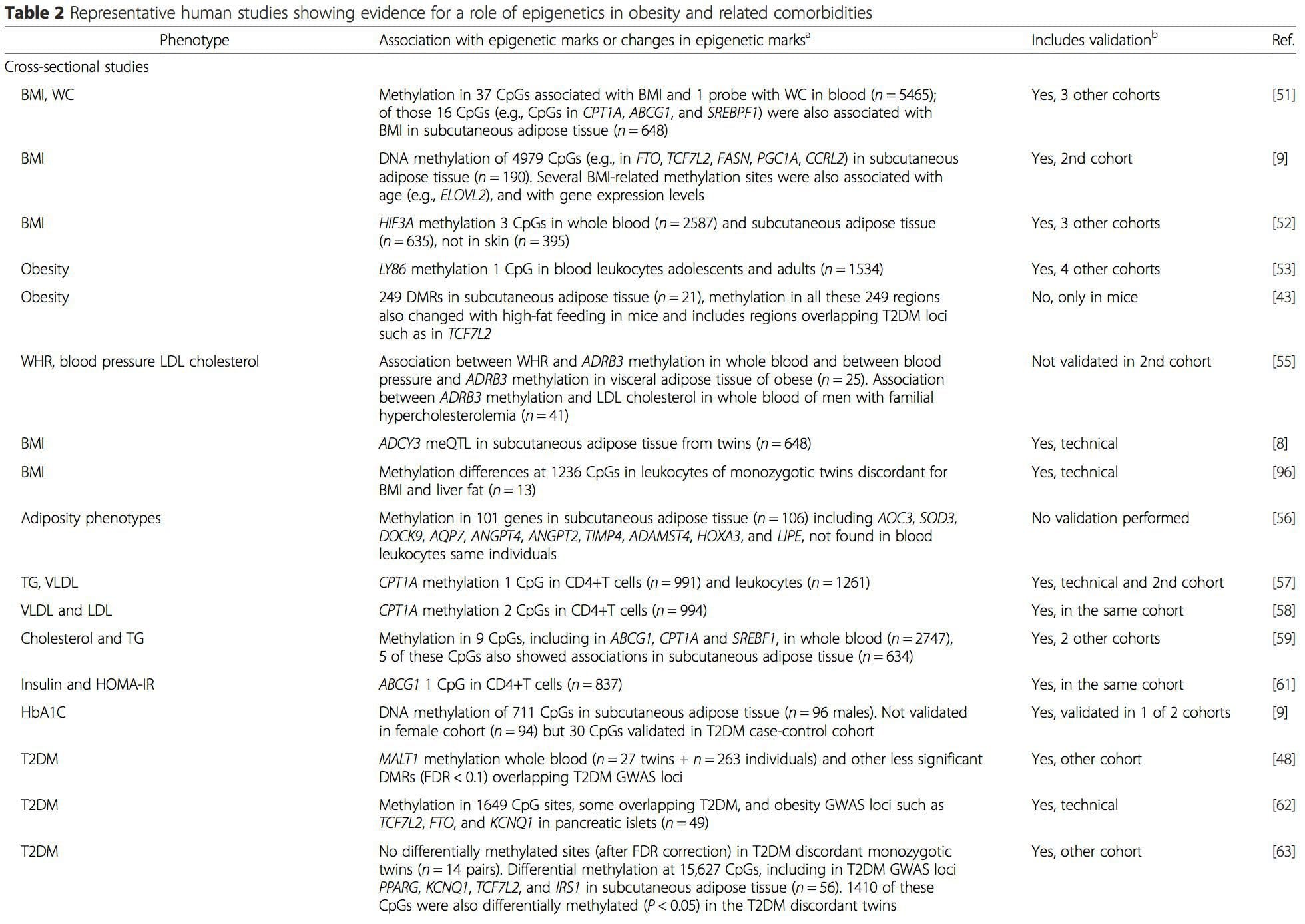
 From these studies, altered methylation of PGC1A, HIF3A, ABCG1, and CPT1A and the previously described RXRA [18] have emerged as biomarkers associated with, or perhaps predictive of, metabolic health that are also plausible candidates for a role in development of metabolic disease.
From these studies, altered methylation of PGC1A, HIF3A, ABCG1, and CPT1A and the previously described RXRA [18] have emerged as biomarkers associated with, or perhaps predictive of, metabolic health that are also plausible candidates for a role in development of metabolic disease.
Interaction Between Genotype And The Epigenome
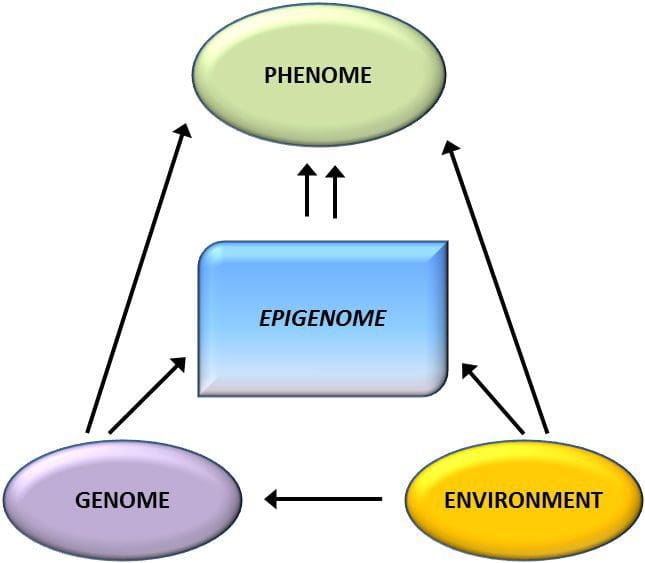 Epigenetic variation is highly influenced by the underlying genetic variation, with genotype estimated to explain ~20–40 % of the variation [6, 8]. Recently, a number of studies have begun to integrate methylome and genotype data to identify methylation quantitative trait loci (meQTL) associated with disease phenotypes. For instance, in adipose tissue, an meQTL overlapping with a BMI genetic risk locus has been identified in an enhancer element upstream of ADCY3 [8]. Other studies have also identified overlaps between known obesity and T2DM risk loci and DMRs associated with obesity and T2DM [43, 48, 62]. Methylation of a number of such DMRs was also modulated by high-fat feeding in mice [43] and weight loss in humans [64]. These results identify an intriguing link between genetic variations linked with disease susceptibility and their association with regions of the genome that undergo epigenetic modifications in response to nutritional challenges, implying a causal relationship. The close connection between genetic and epigenetic variation may signify their essential roles in generating individual variation [65, 66]. However, while these findings suggest that DNA methylation may be a mediator of genetic effects, it is also important to consider that both genetic and epigenetic processes could act independently on the same genes. Twin studies [8, 63, 67] can provide important insights and indicate that inter-individual differences in levels of DNA methylation arise predominantly from non-shared environment and stochastic influences, minimally from shared environmental effects, but also with a significant impact of genetic variation.
Epigenetic variation is highly influenced by the underlying genetic variation, with genotype estimated to explain ~20–40 % of the variation [6, 8]. Recently, a number of studies have begun to integrate methylome and genotype data to identify methylation quantitative trait loci (meQTL) associated with disease phenotypes. For instance, in adipose tissue, an meQTL overlapping with a BMI genetic risk locus has been identified in an enhancer element upstream of ADCY3 [8]. Other studies have also identified overlaps between known obesity and T2DM risk loci and DMRs associated with obesity and T2DM [43, 48, 62]. Methylation of a number of such DMRs was also modulated by high-fat feeding in mice [43] and weight loss in humans [64]. These results identify an intriguing link between genetic variations linked with disease susceptibility and their association with regions of the genome that undergo epigenetic modifications in response to nutritional challenges, implying a causal relationship. The close connection between genetic and epigenetic variation may signify their essential roles in generating individual variation [65, 66]. However, while these findings suggest that DNA methylation may be a mediator of genetic effects, it is also important to consider that both genetic and epigenetic processes could act independently on the same genes. Twin studies [8, 63, 67] can provide important insights and indicate that inter-individual differences in levels of DNA methylation arise predominantly from non-shared environment and stochastic influences, minimally from shared environmental effects, but also with a significant impact of genetic variation.
The Impact Of The Prenatal And Postnatal Environment On The Epigenome
 Prenatal environment: Two recently published studies made use of human populations that experienced “natural” variations in nutrient supply to study the impact of maternal nutrition before or during pregnancy on DNA methylation in the offspring [68, 69]. The first study used a Gambian mother-child cohort to show that both seasonal variations in maternal methyl donor intake during pregnancy and maternal pre-pregnancy BMI were associated with altered methylation in the infants [69]. The second study utilized adult offspring from the Dutch Hunger Winter cohort to investigate the effect of prenatal exposure to an acute period of severe maternal undernutrition on DNA methylation of genes involved in growth and metabolism in adulthood [68]. The results highlighted the importance of the timing of the exposure in its impact on the epigenome, since significant epigenetic effects were only identified in individuals exposed to famine during early gestation. Importantly, the epigenetic changes occurred in conjunction with increased BMI; however, it was not possible to establish in this study whether these changes were present earlier in life or a consequence of the higher BMI.
Prenatal environment: Two recently published studies made use of human populations that experienced “natural” variations in nutrient supply to study the impact of maternal nutrition before or during pregnancy on DNA methylation in the offspring [68, 69]. The first study used a Gambian mother-child cohort to show that both seasonal variations in maternal methyl donor intake during pregnancy and maternal pre-pregnancy BMI were associated with altered methylation in the infants [69]. The second study utilized adult offspring from the Dutch Hunger Winter cohort to investigate the effect of prenatal exposure to an acute period of severe maternal undernutrition on DNA methylation of genes involved in growth and metabolism in adulthood [68]. The results highlighted the importance of the timing of the exposure in its impact on the epigenome, since significant epigenetic effects were only identified in individuals exposed to famine during early gestation. Importantly, the epigenetic changes occurred in conjunction with increased BMI; however, it was not possible to establish in this study whether these changes were present earlier in life or a consequence of the higher BMI.
Other recent studies have provided evidence that prenatal over-nutrition and an obese or diabetic maternal environment are also associated with DNA methylation changes in genes related to embryonic development, growth, and metabolic disease in the offspring [70–73].
While human data are scarce, there are indications that paternal obesity can lead to altered methylation of imprinted genes in the newborn [74], an effect thought to be mediated via epigenetic changes acquired during spermatogenesis.
 Postnatal environment: The epigenome is established de novo during embryonic development, and therefore, the prenatal environment most likely has the most significant impact on the epigenome. However, it is now clear that changes do occur in the “mature” epigenome under the influence of a range of conditions, including aging, exposure to toxins, and dietary alterations. For example, changes in DNA methylation in numerous genes in skeletal muscle and PGC1A in adipose tissue have been demonstrated in response to a high-fat diet [75, 76]. Interventions to lose body fat mass have also been associated with changes in DNA methylation. Studies have reported that the DNA methylation profiles of adipose tissue [43, 64], peripheral blood mononuclear cells [77], and muscle tissue [78] in formerly obese patients become more similar to the profiles of lean subjects following weight loss. Weight loss surgery also partially reversed non-alcoholic fatty liver disease-associated methylation changes in liver [79] and in another study led to hypomethylation of multiple obesity candidate genes, with more pronounced effects in subcutaneous compared to omental (visceral) fat [64]. Accumulating evidence suggests that exercise interventions can also influence DNA methylation. Most of these studies have been conducted in lean individuals [80–82], but one exercise study in obese T2DM subjects also demonstrated changes in DNA methylation, including in genes involved in fatty acid and glucose transport [83]. Epigenetic changes also occur with aging, and recent data suggest a role of obesity in augmenting them [9, 84, 85]. Obesity accelerated the epigenetic age of liver tissue, but in contrast to the findings described above, this effect was not reversible after weight loss [84].
Postnatal environment: The epigenome is established de novo during embryonic development, and therefore, the prenatal environment most likely has the most significant impact on the epigenome. However, it is now clear that changes do occur in the “mature” epigenome under the influence of a range of conditions, including aging, exposure to toxins, and dietary alterations. For example, changes in DNA methylation in numerous genes in skeletal muscle and PGC1A in adipose tissue have been demonstrated in response to a high-fat diet [75, 76]. Interventions to lose body fat mass have also been associated with changes in DNA methylation. Studies have reported that the DNA methylation profiles of adipose tissue [43, 64], peripheral blood mononuclear cells [77], and muscle tissue [78] in formerly obese patients become more similar to the profiles of lean subjects following weight loss. Weight loss surgery also partially reversed non-alcoholic fatty liver disease-associated methylation changes in liver [79] and in another study led to hypomethylation of multiple obesity candidate genes, with more pronounced effects in subcutaneous compared to omental (visceral) fat [64]. Accumulating evidence suggests that exercise interventions can also influence DNA methylation. Most of these studies have been conducted in lean individuals [80–82], but one exercise study in obese T2DM subjects also demonstrated changes in DNA methylation, including in genes involved in fatty acid and glucose transport [83]. Epigenetic changes also occur with aging, and recent data suggest a role of obesity in augmenting them [9, 84, 85]. Obesity accelerated the epigenetic age of liver tissue, but in contrast to the findings described above, this effect was not reversible after weight loss [84].
Collectively, the evidence in support of the capacity to modulate the epigenome in adults suggests that there may be the potential to intervene in postnatal life to modulate or reverse adverse epigenetic programming.
Effect Sizes And Differences Between Tissue Types
 DNA methylation changes associated with obesity or induced by diet or lifestyle interventions and weight loss are generally modest (<15 %), although this varies depending on the phenotype and tissue studied. For instance, changes greater than 20 % have been reported in adipose tissue after weight loss [64] and associations between HIF3A methylation and BMI in adipose tissue were more pronounced than in blood [52].
DNA methylation changes associated with obesity or induced by diet or lifestyle interventions and weight loss are generally modest (<15 %), although this varies depending on the phenotype and tissue studied. For instance, changes greater than 20 % have been reported in adipose tissue after weight loss [64] and associations between HIF3A methylation and BMI in adipose tissue were more pronounced than in blood [52].
The biological relevance of relatively small methylation changes has been questioned. However, in tissues consisting of a mixture of cell types, a small change in DNA methylation may actually reflect a significant change in a specific cell fraction. Integration of epigenome data with transcriptome and other epigenetic data, such as histone modifications, is important, since small DNA methylation changes might reflect larger changes in chromatin structure and could be associated with broader changes in gene expression. The genomic context should also be considered; small changes within a regulatory element such as a promotor, enhancer, or insulator may have functional significance. In this regard, DMRs for obesity, as well as regions affected by prenatal famine exposure and meQTL for metabolic trait loci have been observed to overlap enhancer elements [8, 43, 68]. There is evidence that DNA methylation in famine-associated regions could indeed affect enhancer activity [68], supporting a role of nutrition-induced methylation changes in gene regulation.
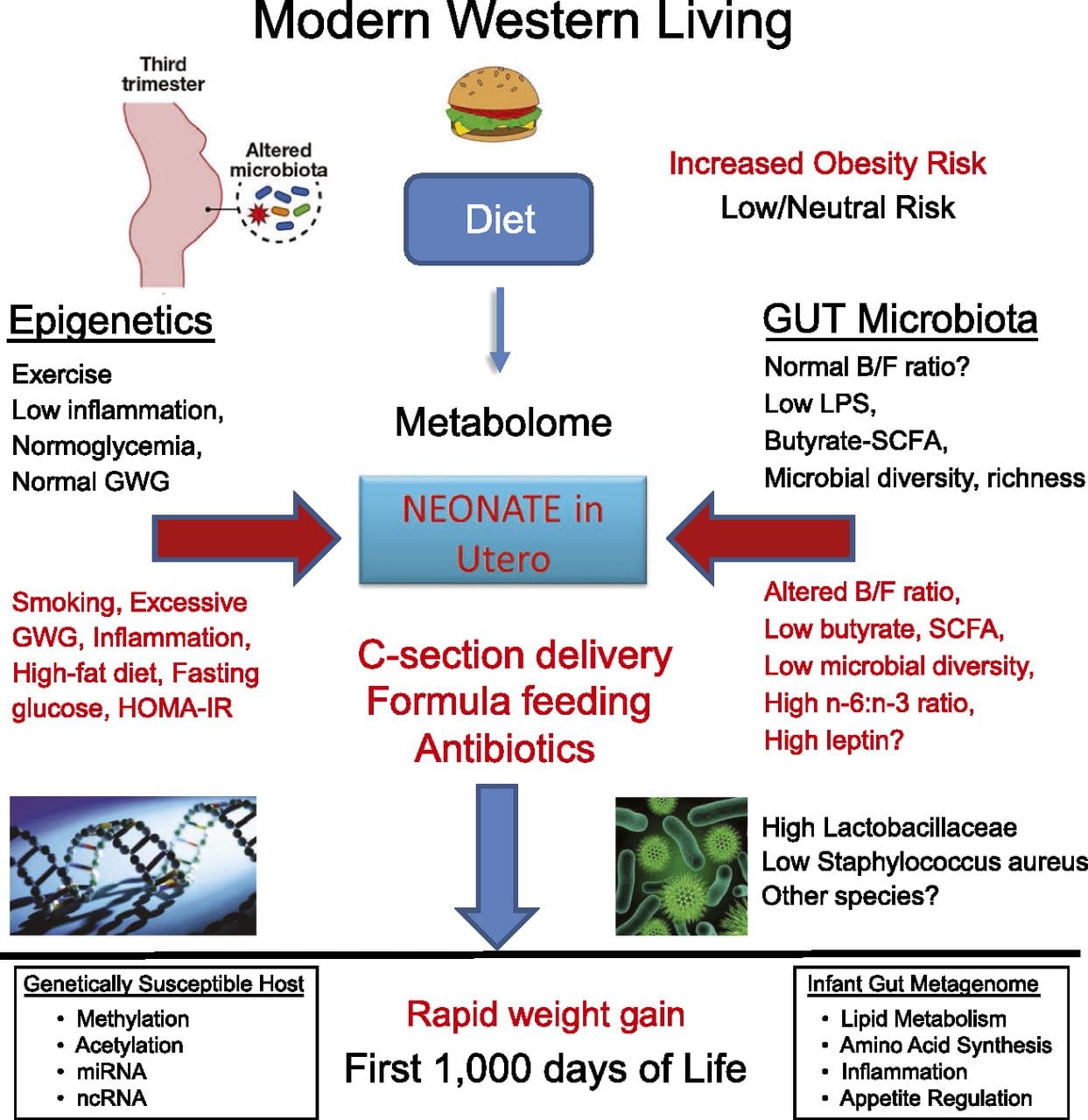
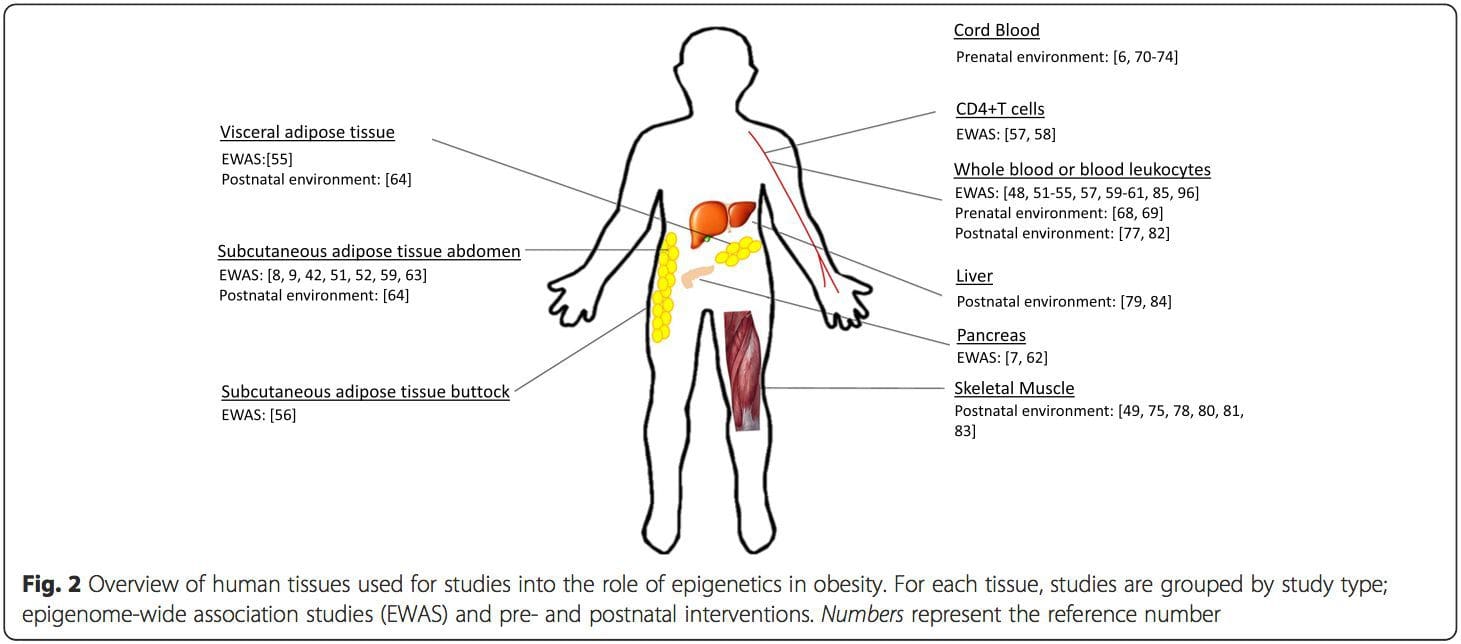
A major limitation in many human studies is that epigenetic marks are often assessed in peripheral blood, rather than in metabolically relevant tissues (Fig. 2). The heterogeneity of blood is an issue, since different cell populations have distinct epigenetic signatures, but algorithms have been developed to estimate the cellular composition to overcome this problem [86]. Perhaps more importantly, epigenetic marks in blood cells may not necessarily report the status of the tissues of primary interest. Despite this, recent studies have provided clear evidence of a relationship between epigenetic marks in blood cells and BMI. In the case of HIF3A for which the level of methylation (beta-value) in the study population ranged from 0.14–0.52, a 10 % increase in methylation was associated with a BMI increase of 7.8 % [52]. Likewise, a 10 % difference in PGC1A methylation may predict up to 12 % difference in fat mass [54].
 Conclusions
Conclusions
The study of the role of epigenetics in obesity and metabolic disease has expanded rapidly in recent years, and evidence is accumulating of a link between epigenetic modifications and metabolic health outcomes in humans. Potential epigenetic biomarkers associated with obesity and metabolic health have also emerged from recent studies. The validation of epigenetic marks in multiple cohorts, the fact that several marks are found in genes with a plausible function in obesity and T2DM development, as well as the overlap of epigenetic marks with known obesity and T2DM genetic loci strengthens the evidence that these associations are real. Causality has so far been difficult to establish; however, regardless of whether the associations are causal, the identified epigenetic marks may still be relevant as biomarkers for obesity and metabolic disease risk.
Effect sizes in easily accessible tissues such as blood are small but do seem reproducible despite variation in ethnicity, tissue type, and analysis methods [51]. Also, even small DNA methylation changes may have biological significance. An integrative “omics” approach will be crucial in further unraveling the complex interactions between the epigenome, transcriptome, genome, and metabolic health. Longitudinal studies, ideally spanning multiple generations, are essential to establishing causal relationships. We can expect more such studies in the future, but this will take time.
While animal studies continue to demonstrate an effect of early life nutritional exposure on the epigenome and metabolic health of the offspring, human data are still limited. However, recent studies have provided clear evidence that exposure to suboptimal nutrition during specific periods of prenatal development is associated with methylation changes in the offspring and therefore have the potential to influence adult phenotype. Animal studies will be important to verify human findings in a more controlled setting, help determine whether the identified methylation changes have any impact on metabolic health, and unravel the mechanisms underlying this intergenerational/transgenerational epigenetic regulation. The identification of causal mechanisms underlying metabolic memory responses, the mode of transmission of the phenotypic effects into successive generations, the degree of impact and stability of the transmitted trait, and the identification of an overarching and unifying evolutionary context also remain important questions to be addressed. The latter is often encapsulated by the predictive adaptive response hypothesis, i.e., a response to a future anticipated environment that increases fitness of the population. However, this hypothesis has increasingly been questioned as there is limited evidence for increased fitness later in life [87].
In summary, outcomes are promising, as the epigenetic changes are linked with adult metabolic health and they act as a mediator between altered prenatal nutrition and subsequent increased risk of poor metabolic health outcomes. New epigenetic marks have been identified that are associated with measures of metabolic health. Integration of different layers of genomic information has added further support to causal relationships, and there have been further studies showing effects of pre- and postnatal environment on the epigenome and health. While many important questions remain, recent methodological advances have enabled the types of large-scale population-based studies that will be required to address the knowledge gaps. The next decade promises to be a period of major activity in this important research area.
Susan J. van Dijk1, Ross L. Tellam2, Janna L. Morrison3, Beverly S. Muhlhausler4,5† and Peter L. Molloy1*†
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed to the drafting and critical revision of the manuscript, and all authors read and approved the final manuscript.
Authors’ information
Beverly S. Muhlhausler and Peter L. Molloy are joint last authors.
Acknowledgements
This work has been supported by a grant from the Science and Industry Endowment Fund (Grant RP03-064). JLM and BSM are supported by the National Health and Medical Research Council Career Development Fellowships (JLM, APP1066916; BSM, APP1004211). We thank Lance Macaulay and Sue Mitchell for critical reading and comments on the manuscript.
Author details
1CSIRO Food and Nutrition Flagship, PO Box 52, North Ryde, NSW 1670, Australia. 2CSIRO Agriculture Flagship, 306 Carmody Road, St Lucia, QLD 4067, Australia. 3Early Origins of Adult Health Research Group, School of Pharmacy and Medical Sciences, Sansom Institute for Health Research, University of South Australia, GPO Box 2471, Adelaide, SA 5001, Australia 4FOODplus Research Centre, Waite Campus, The University of Adelaide, PMB 1, Glen Osmond, SA 5064, Australia. 5Women’s and Children’s Health Research Institute, 72 King William Road, North Adelaide, SA 5006, Australia.
1. WHO. WHO | Overweight and obesity. http://www.who.int/gho/ncd/<br />
risk_factors/overweight/en/index.html. Accessed 29 January 2015.<br />
2. Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery.<br />
Am J Hum Genet. 2012;90:7–24.<br />
3. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic<br />
studies of body mass index yield new insights for obesity biology. Nature.<br />
2015;518:197–206.<br />
4. Ling C, Del Guerra S, Lupi R, Rönn T, Granhall C, Luthman H, et al.<br />
Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and<br />
effect on insulin secretion. Diabetologia. 2008;51:615–22.<br />
5. Van Dijk SJ, Molloy PL, Varinli H, Morrison JL, Muhlhausler BS. Epigenetics<br />
and human obesity. Int J Obes (Lond). 2015;39:85–97.<br />
6. Teh AL, Pan H, Chen L, Ong M-L, Dogra S, Wong J, et al. The effect of<br />
genotype and in utero environment on interindividual variation in neonate<br />
DNA methylomes. Genome Res. 2014;24:1064–74.<br />
7. Olsson AH, Volkov P, Bacos K, Dayeh T, Hall E, Nilsson EA, et al. Genomewide<br />
associations between genetic and epigenetic variation influence<br />
mRNA expression and insulin secretion in human pancreatic islets. PLoS<br />
Genet. 2014;10:e1004735.<br />
8. Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, et al.<br />
Global analysis of DNA methylation variation in adipose tissue from twins<br />
reveals links to disease-associated variants in distal regulatory elements.<br />
Am J Hum Genet. 2013;93:876–90.<br />
9. Ronn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, et al.<br />
Impact of age, BMI and HbA1c levels on the genome-wide DNA<br />
methylation and mRNA expression patterns in human adipose tissue<br />
and identification of epigenetic biomarkers in blood. Hum Mol Genet.<br />
2015;24:3792–813.<br />
10. Waterland RA, Michels KB. Epigenetic epidemiology of the developmental<br />
origins hypothesis. Annu Rev Nutr. 2007;27:363–88.<br />
11. McMillen IC, Rattanatray L, Duffield JA, Morrison JL, MacLaughlin SM, Gentili<br />
S, et al. The early origins of later obesity: pathways and mechanisms. Adv<br />
Exp Med Biol. 2009;646:71–81.<br />
12. Ravelli A, van der Meulen J, Michels R, Osmond C, Barker D, Hales C, et al.<br />
Glucose tolerance in adults after prenatal exposure to famine. Lancet.<br />
1998;351:173–7.<br />
13. McMillen IC, MacLaughlin SM, Muhlhausler BS, Gentili S, Duffield JL,<br />
Morrison JL. Developmental origins of adult health and disease: the role of<br />
periconceptional and foetal nutrition. Basic Clin Pharmacol Toxicol.<br />
2008;102:82–9.<br />
14. Zhang S, Rattanatray L, McMillen IC, Suter CM, Morrison JL. Periconceptional<br />
nutrition and the early programming of a life of obesity or adversity. Prog<br />
Biophys Mol Biol. 2011;106:307–14.<br />
15. Bouret S, Levin BE, Ozanne SE. Gene-environment interactions controlling<br />
energy and glucose homeostasis and the developmental origins of obesity.<br />
Physiol Rev. 2015;95:47–82.<br />
16. Borengasser SJ, Zhong Y, Kang P, Lindsey F, Ronis MJ, Badger TM, et al.<br />
Maternal obesity enhances white adipose tissue differentiation and alters<br />
genome-scale DNA methylation in male rat offspring. Endocrinology.<br />
2013;154:4113–25.<br />
17. Gluckman PD, Lillycrop KA, Vickers MH, Pleasants AB, Phillips ES, Beedle AS,<br />
et al. Metabolic plasticity during mammalian development is directionally<br />
dependent on early nutritional status. Proc Natl Acad Sci U S A.<br />
2007;104:12796–800.<br />
18. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C,<br />
et al. Epigenetic gene promoter methylation at birth is associated with<br />
child’s later adiposity. Diabetes. 2011;60:1528–34.<br />
19. McMillen IC, Adam CL, Muhlhausler BS. Early origins of obesity:<br />
programming the appetite regulatory system. J Physiol. 2005;565(Pt 1):9–17.<br />
20. Begum G, Stevens A, Smith EB, Connor K, Challis JR, Bloomfield F, et al.<br />
Epigenetic changes in fetal hypothalamic energy regulating pathways are<br />
associated with maternal undernutrition and twinning. FASEB J.<br />
2012;26:1694–703.<br />
21. Ge ZJ, Liang QX, Hou Y, Han ZM, Schatten H, Sun QY, et al. Maternal obesity<br />
and diabetes may cause DNA methylation alteration in the spermatozoa of<br />
offspring in mice. Reprod Biol Endocrinol. 2014;12:29.<br />
22. Jousse C, Parry L, Lambert-Langlais S, Maurin AC, Averous J, Bruhat A, et al.<br />
Perinatal undernutrition affects the methylation and expression of the leptin<br />
gene in adults: implication for the understanding of metabolic syndrome.<br />
FASEB J. 2011;25:3271–8.<br />
23. Lan X, Cretney EC, Kropp J, Khateeb K, Berg MA, Penagaricano F, et al.<br />
Maternal diet during pregnancy induces gene expression and DNA<br />
methylation changes in fetal tissues in sheep. Front Genet. 2013;4:49.<br />
24. Li CC, Young PE, Maloney CA, Eaton SA, Cowley MJ, Buckland ME, et al.<br />
Maternal obesity and diabetes induces latent metabolic defects and<br />
widespread epigenetic changes in isogenic mice. Epigenetics. 2013;8:602–11.<br />
25. Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein<br />
restriction of pregnant rats induces and folic acid supplementation prevents<br />
epigenetic modification of hepatic gene expression in the offspring. J Nutr.<br />
2005;135:1382–6.<br />
26. Radford EJ, Ito M, Shi H, Corish JA, Yamazawa K, Isganaitis E, et al. In utero<br />
effects. In utero undernourishment perturbs the adult sperm methylome<br />
and intergenerational metabolism. Science. 2014;345(80):1255903.<br />
27. Suter M, Bocock P, Showalter L, Hu M, Shope C, McKnight R, et al.<br />
Epigenomics: maternal high-fat diet exposure in utero disrupts<br />
peripheral circadian gene expression in nonhuman primates. FASEB J.<br />
2011;25:714–26.<br />
28. Suter MA, Ma J, Vuguin PM, Hartil K, Fiallo A, Harris RA, et al. In utero<br />
exposure to a maternal high-fat diet alters the epigenetic histone code in a<br />
murine model. Am J Obs Gynecol. 2014;210:463 e1–463 e11.<br />
29. Tosh DN, Fu Q, Callaway CW, McKnight RA, McMillen IC, Ross MG, et al.<br />
Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1<br />
mRNA expression and histone structure in rapid vs. delayed postnatal<br />
catch-up growth. Am J Physiol Gastrointest Liver Physiol.<br />
2010;299:G1023–9.<br />
30. Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, Uribe-Lewis S, Ito Y,<br />
et al. Maternal diet and aging alter the epigenetic control of a promoterenhancer<br />
interaction at the Hnf4a gene in rat pancreatic islets. Proc Natl<br />
Acad Sci U S A. 2011;108:5449–54.<br />
31. Braunschweig M, Jagannathan V, Gutzwiller A, Bee G. Investigations on<br />
transgenerational epigenetic response down the male line in F2 pigs. PLoS<br />
One. 2012;7, e30583.<br />
32. Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally<br />
induced transgenerational environmental reprogramming of metabolic<br />
gene expression in mammals. Cell. 2010;143:1084–96.<br />
33. Ost A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, et al. Paternal diet<br />
defines offspring chromatin state and intergenerational obesity. Cell.<br />
2014;159:1352–64.<br />
34. Martínez D, Pentinat T, Ribó S, Daviaud C, Bloks VW, Cebrià J, et al. In utero<br />
undernutrition in male mice programs liver lipid metabolism in the secondgeneration<br />
offspring involving altered Lxra DNA methylation. Cell Metab.<br />
2014;19:941–51.<br />
35. Wei Y, Yang C-R, Wei Y-P, Zhao Z-A, Hou Y, Schatten H, et al. Paternally<br />
induced transgenerational inheritance of susceptibility to diabetes in<br />
mammals. Proc Natl Acad Sci U S A. 2014;111:1873–8.<br />
36. Grossniklaus U, Kelly WG, Kelly B, Ferguson-Smith AC, Pembrey M, Lindquist<br />
S. Transgenerational epigenetic inheritance: how important is it? Nat Rev<br />
Genet. 2013;14:228–35.<br />
37. Pembrey M, Saffery R, Bygren LO. Human transgenerational responses to<br />
early-life experience: potential impact on development, health and<br />
biomedical research. J Med Genet. 2014;51:563–72.<br />
38. Wolff GL, Kodell RL, Moore SR, Cooney CA. Maternal epigenetics and methyl<br />
supplements affect agouti gene expression in Avy/a mice. FASEB J.<br />
1998;12:949–57.<br />
39. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility.<br />
Nat Rev Genet. 2007;8:253–62.<br />
40. Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at<br />
the agouti locus in the mouse. Nat Genet. 1999;23:314–8.<br />
41. Cropley JE, Suter CM, Beckman KB, Martin DI. Germ-line epigenetic<br />
modification of the murine A vy allele by nutritional supplementation. Proc<br />
Natl Acad Sci U S A. 2006;103:17308–12.<br />
42. Hoile SP, Lillycrop KA, Thomas NA, Hanson MA, Burdge GC. Dietary protein<br />
restriction during F0 pregnancy in rats induces transgenerational changes in<br />
the hepatic transcriptome in female offspring. PLoS One. 2011;6, e21668.<br />
43. Multhaup ML, Seldin MM, Jaffe AE, Lei X, Kirchner H, Mondal P, et al. Mousehuman<br />
experimental epigenetic analysis unmasks dietary targets and<br />
genetic liability for diabetic phenotypes. Cell Metab. 2015;21:138–49.<br />
44. Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, et al.<br />
Recommendations for the design and analysis of epigenome-wide<br />
association studies. Nat Methods. 2013;10:949–55.<br />
45. Dayeh TA, Olsson AH, Volkov P, Almgren P, Rönn T, Ling C. Identification of<br />
CpG-SNPs associated with type 2 diabetes and differential DNA methylation<br />
in human pancreatic islets. Diabetologia. 2013;56:1036–46.<br />
46. Relton CL, Davey Smith G. Two-step epigenetic Mendelian randomization: a<br />
strategy for establishing the causal role of epigenetic processes in pathways<br />
to disease. Int J Epidemiol. 2012;41:161–76.<br />
47. Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al.<br />
Epigenome-wide association data implicate DNA methylation as an<br />
intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol.<br />
2013;31:142–7.<br />
48. Yuan W, Xia Y, Bell CG, Yet I, Ferreira T, Ward KJ, et al. An integrated<br />
epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic<br />
twins. Nat Commun. 2014;5:5719.<br />
49. Nitert MD, Dayeh T, Volkov P, Elgzyri T, Hall E, Nilsson E, et al. Impact of an<br />
exercise intervention on DNA methylation in skeletal muscle from firstdegree<br />
relatives of patients with type 2 diabetes. Diabetes. 2012;61:3322–32.<br />
50. Gagnon F, Aïssi D, Carrié A, Morange P-E, Trégouët D-A. Robust validation of<br />
methylation levels association at CPT1A locus with lipid plasma levels.<br />
J Lipid Res. 2014;55:1189–91.<br />
51. Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou Y-H,<br />
et al. Epigenome-wide association atudy (EWAS) of BMI, BMI change, and<br />
waist circumference in African American adults identifies multiple replicated<br />
loci. Hum Mol Genet. 2015:ddv161–.<br />
52. Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aïssi D, Wahl S, et al. DNA<br />
methylation and body-mass index: a genome-wide analysis. Lancet.<br />
2014;6736:1–9.<br />
53. Su S, Zhu H, Xu X, Wang X, Dong Y, Kapuku G, et al. DNA methylation of<br />
the LY86 gene is associated with obesity, insulin resistance, and<br />
inflammation. Twin Res Hum Genet. 2014;17:183–91.<br />
54. Clarke-Harris R, Wilkin TJ, Hosking J, Pinkney J, Jeffery AN, Metcalf BS, et al.<br />
PGC1? promoter methylation in blood at 5–7 years predicts adiposity from<br />
9 to 14 years (EarlyBird 50). Diabetes. 2014;63:2528–37.<br />
55. Guay S-P, Brisson D, Lamarche B, Biron S, Lescelleur O, Biertho L, et al.<br />
ADRB3 gene promoter DNA methylation in blood and visceral adipose<br />
tissue is associated with metabolic disturbances in men. Epigenomics.<br />
2014;6:33–43.<br />
56. Agha G, Houseman EA, Kelsey KT, Eaton CB, Buka SL, Loucks EB. Adiposity is<br />
associated with DNA methylation profile in adipose tissue. Int J Epidemiol.<br />
2014:1–11.<br />
57. Irvin MR, Zhi D, Joehanes R, Mendelson M, Aslibekyan S, Claas SA, et al.<br />
Epigenome-wide association study of fasting blood lipids in the genetics of<br />
lipid-lowering drugs and diet network study. Circulation. 2014;130:565–72.<br />
58. Frazier-Wood AC, Aslibekyan S, Absher DM, Hopkins PN, Sha J, Tsai MY, et al.<br />
Methylation at CPT1A locus is associated with lipoprotein subfraction<br />
profiles. J Lipid Res. 2014;55:1324–30.<br />
59. Pfeifferm L, Wahl S, Pilling LC, Reischl E, Sandling JK, Kunze S, et al. DNA<br />
methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc<br />
Genet. 2015.<br />
60. Petersen A-K, Zeilinger S, Kastenmüller G, Römisch-Margl W, Brugger M, Peters<br />
A, et al. Epigenetics meets metabolomics: an epigenome-wide association<br />
study with blood serum metabolic traits. Hum Mol Genet. 2014;23:534–45.<br />
61. Hidalgo B, Irvin MR, Sha J, Zhi D, Aslibekyan S, Absher D, et al. Epigenomewide<br />
association study of fasting measures of glucose, insulin, and HOMA-IR<br />
in the genetics of lipid lowering drugs and diet network study. Diabetes.<br />
2014;63:801–7.<br />
62. Dayeh T, Volkov P, Salö S, Hall E, Nilsson E, Olsson AH, et al. Genome-wide<br />
DNA methylation analysis of human pancreatic islets from type 2 diabetic<br />
and non-diabetic donors identifies candidate genes that influence insulin<br />
secretion. PLoS Genet. 2014;10, e1004160.<br />
63. Nilsson E, Jansson PA, Perfilyev A, Volkov P, Pedersen M, Svensson MK, et al.<br />
Altered DNA methylation and differential expression of genes influencing<br />
metabolism and inflammation in adipose tissue from subjects with type 2<br />
diabetes. Diabetes. 2014;63:2962–76.<br />
64. Benton MC, Johnstone A, Eccles D, Harmon B, Hayes MT, Lea RA, et al. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight<br />
loss. Gene. 2015;16:1–21.<br />
65. Bateson P, Gluckman P. Plasticity and robustness in development and<br />
evolution. Int J Epidemiol. 2012;41:219–23.<br />
66. Feinberg AP, Irizarry RA, Feinberg AP, Irizarry RA. Evolution in health and<br />
medicine Sackler colloquium: stochastic epigenetic variation as a driving<br />
force of development, evolutionary adaptation, and disease. Proc Natl Acad<br />
Sci U S A. 2010;107(Suppl):1757–64.<br />
67. Martino D, Loke YJ, Gordon L, Ollikainen M, Cruickshank MN, Saffery R, et al.<br />
Longitudinal, genome-scale analysis of DNA methylation in twins from birth<br />
to 18 months of age reveals rapid epigenetic change in early life and pairspecific<br />
effects of discordance. Genome Biol. 2013;14:R42.<br />
68. Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA<br />
methylation signatures link prenatal famine exposure to growth and<br />
metabolism. Nat Commun. 2014;5:5592.<br />
69. Dominguez-Salas P, Moore SE, Baker MS, Bergen AW, Cox SE, Dyer RA, et al.<br />
Maternal nutrition at conception modulates DNA methylation of human<br />
metastable epialleles. Nat Commun. 2014;5:3746.<br />
70. Quilter CR, Cooper WN, Cliffe KM, Skinner BM, Prentice PM, Nelson L, et al.<br />
Impact on offspring methylation patterns of maternal gestational diabetes<br />
mellitus and intrauterine growth restraint suggest common genes and<br />
pathways linked to subsequent type 2 diabetes risk. FASEB J. 2014:1–12.<br />
71. Morales E, Groom A, Lawlor DA, Relton CL. DNA methylation signatures in<br />
cord blood associated with maternal gestational weight gain: results from<br />
the ALSPAC cohort. BMC Res Notes. 2014;7:278.<br />
72. Ruchat SM, Houde AA, Voisin G, St-Pierre J, Perron P, Baillargeon JP, et al.<br />
Gestational diabetes mellitus epigenetically affects genes predominantly<br />
involved in metabolic diseases. Epigenetics. 2013;8:935–43.<br />
73. Liu X, Chen Q, Tsai H-J, Wang G, Hong X, Zhou Y, et al. Maternal<br />
preconception body mass index and offspring cord blood DNA<br />
methylation: exploration of early life origins of disease. Environ Mol<br />
Mutagen. 2014;55:223–30.<br />
74. Soubry A, Murphy SK, Wang F, Huang Z, Vidal AC, Fuemmeler BF, et al.<br />
Newborns of obese parents have altered DNA methylation patterns at<br />
imprinted genes. Int J Obes (Lond). 2015;39:650–7.<br />
75. Jacobsen SC, Brøns C, Bork-Jensen J, Ribel-Madsen R, Yang B, Lara E, et al.<br />
Effects of short-term high-fat overfeeding on genome-wide DNA<br />
methylation in the skeletal muscle of healthy young men. Diabetologia.<br />
2012;55:3341–9.<br />
76. Gillberg L, Jacobsen SC, Rönn T, Brøns C, Vaag A. PPARGC1A DNA<br />
methylation in subcutaneous adipose tissue in low birth weight subjects–<br />
impact of 5 days of high-fat overfeeding. Metabolism. 2014;63:263–71.<br />
77. Huang Y-T, Maccani JZJ, Hawley NL, Wing RR, Kelsey KT, McCaffery JM.<br />
Epigenetic patterns in successful weight loss maintainers: a pilot study. Int J<br />
Obes (Lond). 2015;39:865–8.<br />
78. Barres R, Kirchner H, Rasmussen M, Yan J, Kantor FR, Krook A, Näslund E,<br />
Zierath JR. Weight loss after gastric bypass surgery in human obesity<br />
remodels promoter methylation. Cell Rep. 2013:1–8.<br />
79. Ahrens M, Ammerpohl O, von Schönfels W, Kolarova J, Bens S, Itzel T, et al.<br />
DNA methylation analysis in nonalcoholic fatty liver disease suggests<br />
distinct disease-specific and remodeling signatures after bariatric surgery.<br />
Cell Metab. 2013;18:296–302.<br />
80. Voisin S, Eynon N, Yan X, Bishop DJ. Exercise training and DNA methylation<br />
in humans. Acta Physiol (Oxf). 2014;213:39–59.<br />
81. Lindholm ME, Marabita F, Gomez-Cabrero D, Rundqvist H, Ekström TJ,<br />
Tegnér J, et al. An integrative analysis reveals coordinated reprogramming<br />
of the epigenome and the transcriptome in human skeletal muscle after<br />
training. Epigenetics. 2014;9:1557–69.<br />
82. Denham J, O’Brien BJ, Marques FZ, Charchar FJ. Changes in the leukocyte<br />
methylome and its effect on cardiovascular related genes after exercise.<br />
J Appl Physiol. 2014:jap.00878.2014.<br />
83. Rowlands DS, Page RA, Sukala WR, Giri M, Ghimbovschi SD, Hayat I, et al.<br />
Multi-omic integrated networks connect DNA methylation and miRNA with<br />
skeletal muscle plasticity to chronic exercise in type 2 diabetic obesity.<br />
Physiol Genomics. 2014;46:747–65.<br />
84. Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M,<br />
et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad<br />
Sci. 2014;111:15538–43.<br />
85. Almén MS, Nilsson EK, Jacobsson JA, Kalnina I, Klovins J, Fredriksson R, et al.<br />
Genome-wide analysis reveals DNA methylation markers that vary with<br />
both age and obesity. Gene. 2014.;548:61–7<br />
86. Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments<br />
in analysis of DNA methylation data. Bioinformatics. 2014;30:1431–9.<br />
87. Wells JC. A critical appraisal of the predictive adaptive response hypothesis.<br />
Int J Epidemiol. 2012;41:229–35.<br />
88. Williams-Wyss O, Zhang S, MacLaughlin SM, Kleemann D, Walker SK, Suter<br />
CM, et al. Embryo number and periconceptional undernutrition in the<br />
sheep have differential effects on adrenal epigenotype, growth, and<br />
development. Am J Physiol Endocrinol Metab. 2014;307:E141–50.<br />
89. Zhang S, Rattanatray L, Morrison JL, Nicholas LM, Lie S, McMillen IC.<br />
Maternal obesity and the early origins of childhood obesity: weighing up<br />
the benefits and costs of maternal weight loss in the periconceptional<br />
period for the offspring. Exp Diabetes Res. 2011;2011:585749.<br />
90. Zhang S, Williams-Wyss O, MacLaughlin SM, Walker SK, Kleemann DO, Suter<br />
CM, et al. Maternal undernutrition during the first week after conception<br />
results in decreased expression of glucocorticoid receptor mRNA in the<br />
absence of GR exon 17 hypermethylation in the fetal pituitary in late<br />
gestation. J Dev Orig Heal Dis. 2013;4:391–401.<br />
91. Lie S, Morrison JL, Williams-Wyss O, Suter CM, Humphreys DT, Ozanne SE,<br />
et al. Periconceptional undernutrition programs changes in insulin-signaling<br />
molecules and microRNAs in skeletal muscle in singleton and twin fetal<br />
sheep. Biol Reprod. 2014;90:5.<br />
92. Van Straten EM, van Meer H, Huijkman NC, van Dijk TH, Baller JF, Verkade<br />
HJ, et al. Fetal liver X receptor activation acutely induces lipogenesis but<br />
does not affect plasma lipid response to a high-fat diet in adult mice. Am J<br />
Physiol Endocrinol Metab. 2009;297:E1171–8.<br />
93. Fernandez-Twinn DS, Alfaradhi MZ, Martin-Gronert MS, Duque-Guimaraes<br />
DE, Piekarz A, Ferland-McCollough D, et al. Downregulation of IRS-1 in<br />
adipose tissue of offspring of obese mice is programmed cellautonomously<br />
through post-transcriptional mechanisms. Mol Metab.<br />
2014;3:325–33.<br />
94. Waterland RA, Travisano M, Tahiliani KG. Diet-induced hypermethylation at<br />
agouti viable yellow is not inherited transgenerationally through the female.<br />
FASEB J. 2007;21:3380–5.<br />
95. Ge ZJ, Luo SM, Lin F, Liang QX, Huang L, Wei YC, et al. DNA methylation in<br />
oocytes and liver of female mice and their offspring: effects of high-fat-dietinduced<br />
obesity. Env Heal Perspect. 2014;122:159–64.<br />
96. Ollikainen M, Ismail K, Gervin K, Kyllönen A, Hakkarainen A, Lundbom J, et al.<br />
Genome-wide blood DNA methylation alterations at regulatory elements<br />
and heterochromatic regions in monozygotic twins discordant for obesity<br />
and liver fat. Clin Epigenetics. 2015;7:1–13.
Post Disclaimer
General Disclaimer, Licenses and Board Certifications *
Professional Scope of Practice *
The information herein on "The Role Of Epigenetics In Obesity And Metabolic Disease" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a Multi-State board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our multidisciplinary team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those on this site and on our family practice-based chiromed.com site, focusing on naturally restoring health for patients of all ages.
Our areas of multidisciplinary practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is multidisciplinary, focusing on musculoskeletal and physical medicine; wellness; contributing etiological viscerosomatic disturbances within clinical presentations; associated somato-visceral reflex clinical dynamics; subluxation complexes; sensitive health issues; and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and licensure jurisdiction. We use functional health & wellness protocols to treat and support care for musculoskeletal injuries or disorders.
Our videos, posts, topics, and insights address clinical matters and issues that directly or indirectly relate to our clinical scope of practice.
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies upon request to regulatory boards and the public.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Multidisciplinary Licensing & Board Certifications:
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License #: TX5807, Verified: TX5807
New Mexico DC License #: NM-DC2182, Verified: NM-DC2182
Multi-State Advanced Practice Registered Nurse (APRN*) in Texas & Multi-States
Multi-state Compact APRN License by Endorsement (42 States)
Texas APRN License #: 1191402, Verified: 1191402 *
New Mexico CNP License#: 90560, Verified
Florida APRN License #: 11043890, Verified: APRN11043890 *
Colorado License #: C-APN.0105610-C-NP, Verified: C-APN.0105610-C-NP
New York License #: N25929, Verified N25929
License Verification Link: Nursys License Verifier
* Prescriptive Authority Authorized
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
| Yes | 363LF0000X - Nurse Practitioner - Family | NM |
90560 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
📆 Schedule Appointment: Schedule 24/7 (Click Here)